
An official website of the United States government
The .gov means it's official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you're on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
- Browse Titles
NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-.


StatPearls [Internet].
Thalassemia.
Hamza Bajwa ; Hajira Basit .
Affiliations
Last Update: August 8, 2023 .
- Continuing Education Activity
Thalassemia is a heterogeneous group of blood disorders affecting the hemoglobin genes and resulting in ineffective erythropoiesis. The decreased production of hemoglobin results in anemia in early age and frequent blood transfusions are required to keep up the hemoglobin levels. This activity outlines the evaluation and treatment of thalassemia and highlights the role of an interprofessional team in managing the patients with this condition.
- Summarize the etiology of thalassemias.
- Review of different laboratory and bedside evaluation techniques in the management of thalassemia patients.
- Outline the treatment and management options available for thalassemia.
- Identify interprofessional team strategies for improving care coordination and communication to improve outcomes in thalassemia.
- Introduction
Thalassemias are a heterogeneous grouping of genetic disorders that result from a decreased synthesis of alpha or beta chains of hemoglobin (Hb). Hemoglobin serves as the oxygen-carrying component of the red blood cells. It consists of two proteins, an alpha, and a beta. If the body does not manufacture enough of one or the other of these two proteins, the red blood cells do not form correctly and cannot carry sufficient oxygen; this causes anemia that begins in early childhood and lasts throughout life. Thalassemia is an inherited disease, meaning that at least one of the parents must be a carrier for the disease. It is caused by either a genetic mutation or a deletion of certain key gene fragments.
Alpha thalassemia is caused by alpha-globin gene deletion which results in reduced or absent production of alpha-globin chains. Alpha globin gene has 4 alleles and disease severity ranges from mild to severe depending on the number of deletions of the alleles. Four allele deletion is the most severe form in which no alpha globins are produced and the excess gamma chains (present during the fetal period) form tetramers. It is incompatible with life and results in hydrops fetalis. One allele deletion is the mildest form and is mostly clinically silent.
Beta thalassemia results from point mutations in the beta-globin gene. It is divided into three categories based on the zygosity of the beta-gene mutation. A heterozygous mutation (beta-plus thalassemia) results in beta-thalassemia minor in which beta chains are underproduced. It is mild and usually asymptomatic. Beta thalassemia major is caused by a homozygous mutation (beta-zero thalassemia) of the beta-globin gene, resulting in the total absence of beta chains. It manifests clinically as jaundice, growth retardation, hepatosplenomegaly, endocrine abnormalities, and severe anemia requiring life-long blood transfusions. The condition in between these two types is called beta-thalassemia intermedia with mild to moderate clinical symptoms.
- One mutated gene: Mild signs and symptoms. The condition is called thalassemia minor.
- Two mutated genes: Signs and symptoms will be moderate to severe. This condition is called thalassemia major, or Cooley anemia. Babies born with two mutated beta hemoglobin genes are usually healthy at birth but disease starts to manifest after 6 months of life when fetal hemoglobin (Hb-gamma) disappears and is replaced by adult Hb.
The excess unpaired alpha-globin chains in beta-thalassemia aggregate and form precipitates that damage red cell membranes and result in intravascular hemolysis. This premature death of erythroid precursor cells leads to ineffective erythropoiesis and later results in extramedullary expansion of hematopoiesis.
Coinheritance of alpha thalassemia: Beta-thalassemia patients with coinheritance of alpha thalassemia have a milder clinical course due to a less severe alpha-beta chain imbalance.
Coexistence of sickle cell trait: The presence of sickle cell trait with beta-thalassemia is a major hemoglobinopathy and results in manifestations of sickle cell disease. Unlike sickle cell trait in which major Hb is HbA, in the co-existence state the major Hb is HbS which constitutes more than 60% of Hb depending on the nature of the disease (beta-zero or beta-plus0.)
Hemoglobin (HbE) is also a common Hb variant found in Southeast Asia population. It has a correlation with a beta-thalassemia phenotype, as people with thalassemia in this territory are commonly found to have HbE.
Two new terminologies being used more often in clinical settings are transfusion requiring and non-transfusion requiring thalassemias and all the basic classification falls into these two types depending on the requirement of frequent blood transfusions or not. [1] [2] [3]
Thalassemia is autosomal recessive, which means both the parents must be affected with or carriers for the disease to transfer it to the next generation. It is caused by mutations or deletions of the Hb genes, resulting in underproduction or absence of alpha or beta chains. There are over 200 mutations identified as the culprits for causing thalassemias. Alpha thalassemia is caused by deletions of alpha-globin genes, and beta thalassemias are caused by a point mutation in splice site and promoter regions of the beta-globin gene on chromosome 11. [4]
- Epidemiology
Alpha thalassemia is prevalent in Asian and African populations while beta-thalassemia is more prevalent in the Mediterranean population, although it is relatively common in Southeast Asia and Africa too. Prevalence in these regions may be as high as 10%. The true numbers of thalassemia affected patients in the United States are unknown, as there is no effective screening method in place. [4]
- History and Physical
Thalassemia presentation varies widely depending on the type and severity. A complete history and physical examination can give several clues that are sometimes not obvious to the patient themselves. The following findings can be noted:
Skin can show pallor due to anemia and jaundice due to hyperbilirubinemia resulting from intravascular hemolysis. Patients usually report fatigue due to anemia as the first presenting symptom. Extremities examination can show ulcerations. Chronic iron deposition due to multiple transfusions can result in bronze skin.
Musculoskeletal
Extramedullary expansion of hematopoiesis results in deformed facial and other skeletal bones and an appearance known as chipmunk face.
Iron deposition in cardiac myocytes due to chronic transfusions can disrupt the cardiac rhythm, and the result is various arrhythmias. Due to chronic anemia, overt heart failure can also result.
Chronic hyperbilirubinemia can lead to precipitation of bilirubin gall stones and manifest as typical colicky pain of cholelithiasis. Hepatosplenomegaly can result from chronic iron deposition and also from extramedullary hematopoiesis in these organs. Splenic infarcts or autophagy result from chronic hemolysis due to poorly regulated hematopoiesis.
Hepatic involvement is a common finding in thalassemias, particularly due to the chronic need for transfusions. Chronic liver failure or cirrhosis can result from chronic iron deposition or transfusion-related viral hepatitis.
Slow Growth Rates
Anemia can inhibit a child's growth rate, and thalassemia can cause a delay in puberty. Particular attention should focus on the child's growth and development according to age.
Endocrinopathies
Iron overload can lead to its deposition in various organ systems of the body and resultant decreased functioning of the respective systems. The deposition of iron in the pancreas can lead to diabetes mellitus; in the thyroid or parathyroid glands can lead to hypothyroidism and hypoparathyroidism, respectively. The deposition in joints leads to chronic arthropathies. In the brain, iron prefers to accumulate in the substantia nigra and manifests as early-onset Parkinson's disease and various other physiatry problems. These symptoms fall in the vast kingdom of hemochromatosis. [5]
Several laboratory tests have been developed to screen and diagnose thalassemia:
Complete blood count (CBC): CBC is often the first investigation in a suspected case of thalassemia. A CBC showing low hemoglobin and low MCV is the first indication of thalassemia, after ruling out iron deficiency as the cause of anemia. The calculation of the Mentzer index (mean corpuscular volume divided by red cell count) is useful. A Mentzer lower than 13 suggests that the patient has thalassemia, and an index of more than 13 suggests that the patient has anemia due to iron deficiency. [6]
Peripheral blood smear: A blood smear (also called peripheral smear and manual differential) is next, to assess additional red cell properties. Thalassemia can present with the following findings on the peripheral blood smear:
- Microcytic cells (low MCV)
- Hypochromic cells
- Variation in size and shape (anisocytosis and poikilocytosis)
- Increased percentage of reticulocytes
- Target cells
- Heinz bodies
Iron studies (serum iron, ferritin, unsaturated iron-binding capacity (UIBC), total iron-binding capacity (TIBC), and percent saturation of transferrin) are also done to rule out iron deficiency anemia as the underlying cause.
Erythrocyte porphyrin levels may be checked to distinguish an unclear beta-thalassemia minor diagnosis from iron deficiency or lead poisoning. Individuals with beta-thalassemia will have normal porphyrin levels, but those with the latter conditions will have elevated porphyrin levels.
Hemoglobin electrophoresis: Hemoglobinopathy (Hb) evaluation assesses the type and relative amounts of hemoglobin present in red blood cells. Hemoglobin A (HbA), composed of both alpha and beta-globin chains, is the type of hemoglobin that typically makes up 95% to 98% of hemoglobin for adults. Hemoglobin A2 (HbA2) is normally 2% to 3% of hemoglobin, while hemoglobin F usually makes up less than 2% of hemoglobin in adults.
Beta thalassemia disturbs the balance of beta and alpha hemoglobin chain formation. Patients with the beta-thalassemia major usually have larger percentages of HbF and HbA2 and absent or very low HbA. Those with beta-thalassemia minor usually have a mild elevation of HbA2 and mild decrease of HbA. HbH is a less common form of hemoglobin that may be seen in some cases of alpha thalassemia. HbS is the hemoglobin prevalent in people with sickle cell disease.
Hemoglobinopathy (Hb) assessment is used for prenatal screening when parents are at high risk for hemoglobin abnormalities and state-mandated newborn hemoglobin screening.
DNA analysis: These tests serve to help confirm mutations in the alpha and beta globin-producing genes. DNA testing is not a routine procedure but can be used to help diagnose thalassemia and to determine carrier status if needed.
Since having relatives carrying mutations for thalassemia increases a person's risk of carrying the same mutant gene, family studies may be necessary to assess carrier status and the types of mutations present in other family members.
Genetic testing of amniotic fluid is useful in those rare instances where a fetus has an increased risk for thalassemia. This is particularly important if both parents likely carry a mutation because that increases the risk that their child may inherit a combination of abnormal genes, causing a more severe form of thalassemia. Prenatal diagnosis with chorionic villi sampling at 8 to 10 weeks or by amniocentesis at 14 to 20 weeks’ gestation can be carried out in high-risk families. [7] [6]
Multisystem evaluation: Evaluation of all related systems should be done on a regular basis due to their frequent involvement in the disease progression. Biliary tract and gall bladder imaging, abdominal ultrasonography, cardiac MRI, serum hormone measurements are a few examples that can be done or repeated depending on the clinical suspicion and case description.
- Treatment / Management
Thalassemia treatment depends on the type and severity of the disease.
Mild thalassemia (Hb: 6 to 10g/dl):
Signs and symptoms are generally mild with thalassemia minor and little if any, treatment is needed. Occasionally, patients may need a blood transfusion, particularly after surgery, following childbirth, or to help manage thalassemia complications.
Moderate to severe thalassemia (Hb less than 5 to 6g/dl):
- Frequent blood transfusions: More severe forms of thalassemia often require regular blood transfusions, possibly every few weeks. The goal is to maintain Hb at around 9 to 10 mg/dl to give the patients a sense of well being and also to keep a check on erythropoiesis and suppress extramedullary hematopoiesis. To limit transfusion-related complications, washed, packed red blood cells (RBCs) at approximately 8 to 15 mL cells per kilogram (kg) of body weight over 1 to 2 hours are recommended.
- Chelation therapy: Due to chronic transfusions, iron starts to get deposited in various organs of the body. Iron chelators (deferasirox, deferoxamine, deferiprone) are given concomitantly to remove extra iron from the body.
- Stem cell transplant: Stem cell transplant, (bone marrow transplant), is a potential option in selected cases, such as children born with severe thalassemia. It can eliminate the need for lifelong blood transfusions. [8] However, this procedure has its own complications, and the clinician must weigh these against the benefits. Risks include including graft vs. host disease, chronic immunosuppressive therapy, graft failure, and transplantation-related mortality. [9]
- Gene therapy: It is the latest advancement in severe thalassemia management. It involves harvesting the autologous hematopoietic stem cells (HSCs) from the patient and genetically modifying them with vectors expressing the normal genes. These are then reinfused to the patients after they have undergone the required conditioning to destroy the existing HSCs. The genetically modified HSCs produce normal hemoglobin chains, and normal erythropoiesis ensues.
- Genome editing techniques: Another recent approach is editing genomic libraries, such as zinc-finger nucleases, transcription activator-like effectors, and cluster regulated interspaced short palindromic repeats (CRISPR) with Cas9 nuclease system. These techniques target specific mutation sites and replace them with the normal sequence. The limitation of this technique is to produce a large number of corrected genes sufficient to cure the disease. [10]
- Splenectomy: Patients with thalassemia major often undergo splenectomy to limit the number of required transfusions. Splenectomy is the usual recommendation when the annual transfusion requirement increases to or more than 200 to 220 mL RBCs/kg/year with a hematocrit value of 70%. Splenectomy not only limits the number of required transfusions but also controls the spread of extramedullary hematopoiesis. Postsplenectomy immunizations are necessary to prevent bacterial infections, including Pneumococcus , Meningococcus , and Haemophilus influenzae . Postsplenectomy sepsis is possible in children, so this procedure is deferred until 6 to 7 years of age, and then penicillin is given for prophylaxis until they reach a certain age.
- Cholecystectomy : Patients can develop cholelithiasis due to increased Hb breakdown and bilirubin deposition in the gallbladder. If it becomes symptomatic, patients should undergo cholecystectomy at the same time when they are undergoing splenectomy.
Diet and exercise:
Reports exist that drinking tea aids in reducing iron absorption from the intestinal tract. So, in thalassemia patients tea might be a healthy drink to use routinely. Vitamin C helps in iron excretion from the gut, especially when used with deferoxamine. But using vitamin C in large quantities and without concomitant deferoxamine use, there is a higher risk for fatal arrhythmias. So, the recommendation is to use low quantities of vitamin C along with iron chelators (deferoxamine). [10]
- Differential Diagnosis
- Iron deficiency anemia: This is ruled out by iron studies and Mentzer index.
- Anemia of chronic disease and renal failure: Elevated markers of inflammation (CRP, ESR) point in this direction.
- Sideroblastic anemias: These are ruled out by iron studies and peripheral blood smear.
- Lead poisoning: This is ruled out by measuring serum protoporphyrin level.
Thalassemia minor is usually asymptomatic and has a good prognosis. It normally does not increase morbidity or mortality.
Thalassemia major is a severe disease, and the long-term prognosis depends on the treatment adherence to transfusion and iron chelation therapies. [11]
- Complications
Thalassemia major can produce the following complications [12] [13] :
- Jaundice and gall stones due to hyperbilirubinemia
- Cortical thinning and distortion of bones due to extramedullary hematopoiesis
- High output cardiac failure due to severe anemia, cardiomyopathies, and arrhythmias - cardiac involvement is the major cause of mortality in thalassemia patients
- Hepatosplenomegaly due to extramedullary hematopoiesis and excess iron deposition due to repeated blood transfusions
- Excess iron can lead to findings of primary hemochromatosis such as endocrine abnormalities, joint problems, skin discoloration, etc.
- Neurological complications such as peripheral neuropathies
- Slow growth rate and delayed puberty
- Increased risk of parvovirus B19 infection
- Deterrence and Patient Education
Patients should be educated to keep a check on their disease by following an appropriate treatment plan and adopting healthy living habits.
- Avoid excess iron. Unless the doctor recommends otherwise, patients should avoid multivitamins or other supplements that contain iron.
- Eat a healthy diet. Eating a balanced diet that contains plenty of nutritious foods can help the patient feel better and boost energy. Doctors sometimes also recommend taking a folic acid supplement to help make new red blood cells.
- Avoid infections. Patients should try maximally to protect themselves from infections, especially following a splenectomy. An annual flu shot, meningitis, pneumococcal, and hepatitis B vaccines are recommended to prevent infections.
Patients should also receive education about the hereditary nature of the disease. If both parents have thalassemia minor, there is a 1/4th chance that they will have a child with thalassemia major. If one parent has beta-thalassemia minor and the other parent has some form of beta-globin gene defect, i.e., sickle cell defect, they should also be counseled about the possibility of disease transfer to their children. Patients with thalassemias should understand that their disease is not due to iron deficiency and that iron supplements will not cure the anemia; in fact, it will lead to more iron buildup if they are already receiving blood transfusions. [14]
- Enhancing Healthcare Team Outcomes
Thalassemia has negative repercussions for many organs, and without a cure, it has high morbidity. The disorder is best managed by an interprofessional team that includes a thalassemia care team, cardiologist, hepatologist, endocrinologist, and psychologist. Also, family care, nursing support, and social support are an integral part of the management. A lead consultant should be in charge of the patient care, and a nurse specialist, along with other specialists in the respective fields, should be involved to cover all the aspects of the disease. Patient education is crucial, and social worker involvement, including a geneticist, is essential. In some parts of the world, preventive strategies include prenatal screening, restrictions on issuing marriage licenses to two people with the same disease. The screening of children and pregnant women who visit clinicians is an effective strategy to limit the disease morbidity. The social worker should ensure that the caregiver/patient has adequate support and financial resources so that they can continue with treatment. Nurses should educate patients on the importance of treatment compliance to avoid serious complications, as well as monitoring treatment progress. Pharmacists may soon play a greater role as there are new drug products to assist in gene therapy on the horizon that can eliminate the need for ongoing transfusions.
Active collaboration and discussion between interprofessional team members help in the better understanding of the progression or control of the disease.[Level 5]
- Review Questions
- Access free multiple choice questions on this topic.
- Comment on this article.
Electrophoresis Patterns in Beta-Thalassemia Modified from: Wilson et al. Chapter 11 Evaluation of Anemia, Leukopenia, and Thrombocytopenia. In Jaffe et al: Hematopathology. 2nd ed. Elsevier Health Sciences. Pg197, 2011.
Peripheral blood with features of beta-thalassemia minor. Microcytosis and frequent target cells are characteristic. Contributed by David T Lynch, MD
Peripheral blood picture of beta thalassemia major patient showing hypochromic, microcytic red blood cells along with target cells. Contributed by Hamza Bajwa, MD
Disclosure: Hamza Bajwa declares no relevant financial relationships with ineligible companies.
Disclosure: Hajira Basit declares no relevant financial relationships with ineligible companies.
This book is distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0) ( http://creativecommons.org/licenses/by-nc-nd/4.0/ ), which permits others to distribute the work, provided that the article is not altered or used commercially. You are not required to obtain permission to distribute this article, provided that you credit the author and journal.
- Cite this Page Bajwa H, Basit H. Thalassemia. [Updated 2023 Aug 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-.
In this Page
Bulk download.
- Bulk download StatPearls data from FTP
Related information
- PMC PubMed Central citations
- PubMed Links to PubMed
Similar articles in PubMed
- Review Alpha-Thalassemia. [GeneReviews(®). 1993] Review Alpha-Thalassemia. Tamary H, Dgany O. GeneReviews(®). 1993
- Review Beta-Thalassemia. [GeneReviews(®). 1993] Review Beta-Thalassemia. Langer AL. GeneReviews(®). 1993
- Review Sickle Cell Disease. [GeneReviews(®). 1993] Review Sickle Cell Disease. Bender MA, Carlberg K. GeneReviews(®). 1993
- Review Beta-thalassemia. [Orphanet J Rare Dis. 2010] Review Beta-thalassemia. Galanello R, Origa R. Orphanet J Rare Dis. 2010 May 21; 5:11. Epub 2010 May 21.
- Review Laboratory investigation of hemoglobinopathies and thalassemias: review and update. [Clin Chem. 2000] Review Laboratory investigation of hemoglobinopathies and thalassemias: review and update. Clarke GM, Higgins TN. Clin Chem. 2000 Aug; 46(8 Pt 2):1284-90.
Recent Activity
- Thalassemia - StatPearls Thalassemia - StatPearls
Your browsing activity is empty.
Activity recording is turned off.
Turn recording back on
Connect with NLM
National Library of Medicine 8600 Rockville Pike Bethesda, MD 20894
Web Policies FOIA HHS Vulnerability Disclosure
Help Accessibility Careers
- Reference Manager
- Simple TEXT file
People also looked at
Systematic review article, new insights into pathophysiology of β-thalassemia.

- 1 Hematology Service, Virgen de la Arrixaca University Hospital, Murcia, Spain
- 2 Biomedical Research Institute of Murcia (IMIB), Murcia, Spain
- 3 Centro de Investigaci3n Biomédica en Red de Enfermedades Raras (CIBERER), Madrid, Spain
β-thalassemia is a disease caused by genetic mutations including a nucleotide change, small insertions or deletions in the β-globin gene, or in rare cases, gross deletions into the β-globin gene. These mutations affect globin-chain subunits within the hemoglobin tetramer what induces an imbalance in the α/β-globin chain ratio, with an excess of free α-globin chains that triggers the most important pathogenic events of the disease: ineffective erythropoiesis, chronic anemia/chronic hypoxia, compensatory hemopoietic expansion and iron overload. Based on advances in our knowledge of the pathophysiology of β-thalassemia, in recent years, emerging therapies and clinical trials are being conducted and are classified into three major categories based on the different approach features of the underlying pathophysiology: correction of the α/β-globin disregulation; improving iron overload and reverse ineffective erythropoiesis. However, pathways such as the dysregulation of transcriptional factors, activation of the inflammasome, or approach to mechanisms of bone mineral loss, remain unexplored for future therapeutic targets. In this review, we update the main pathophysiological pathways involved in β-thalassemia, focusing on the development of new therapies directed at new therapeutic targets.
Introduction
Thalassemias is an inherited hemoglobin disorder characterized by reduced or absent globin chain synthesis, resulting in variable clinical phenotypes from severe chronic anemia requiring lifelong transfusion and iron chelating therapy to asymptomatic individuals ( 1 ).
Traditionally, β-thalassemias have been more common in countries in the Mediterranean area, North and Central Africa, Southeast Asia, and the Middle East. However, as a result of migrations of populations, β-thalassemias are now encountered in other regions, such as Northern Europe and North America ( 2 ).
β -thalassemia has a broad clinical spectrum, and traditionally has been classified in the clinic in thalassemia major (TM), thalassemia intermedia (TI), and thalassemia minor ( Figure 1 ). The TM grouped patients with more severe anemia from an early age who require periodic blood transfusions associated with iron chelation for life, while thalassemia minor, the less severe manifestation, is characterized by people with mild asymptomatic anemia and a heterozygous condition (trait) for thalassemia. TI constituted a group with a variable clinical spectrum, from mild to moderate to moderately severe anemia, who do not require blood transfusions on a regular basis, sometimes only occasionally, but who do develop various complications of thalassemia such as extramedullary hematopoiesis, pulmonary hypertension, iron overload, leg ulcers, skeletal deformities, and growth retardation ( 3 , 4 ).

Figure 1 . Types of thalassemia. Genotype–Phenotype Association. α and β-thalassemias are genetically heterogeneous diseases. The clinical management with RBC transfusions is an essential factor in classifying them as either transfusion-dependent thalassemia (TDT) or non–transfusion-dependent thalassemia (NTDT). Patients with TDT need life-long regular transfusions for survival in early childhood while patients with NTDT do not need life-long regular transfusions for survival and normally have later in childhood or even in adulthood with mild/moderate anemia that requires only occasional or short-course regular transfusions under concrete clinical circumstances during times of erythroid stress (infection, pregnancy, surgery, or aplastic crisis); however, usually present the typical complications of TDT such as extramedullary hematopoiesis, iron overload, leg ulcers, and osteoporosis. Patients with TDT include those with β-thalassemia major or severe forms of β-thalassemia intermedia, HbE/β-thalassemia, or α-thalassemia/HbH disease. NTDT mainly encompasses three clinically distinct forms: β-thalassemia intermedia (β-TI), hemoglobin E/β- thalassemia (mild and moderate forms), and α-thalassemia intermedia (hemoglobin H disease).
Recently, this classification has changed due to better understanding of the pathophysiology of the disease and findings focused on the clinical management and complications of IT that show that these patients may present with the same serious complications as transfused patients later in life. In 2012, the International Thalassemia Federation adopted the new terminology for clinical classification of transfusion-dependent thalassemia (TDT) and non–transfusion-dependent thalassemia (NTDT) that groups in three different types in the clinic: α-thalassemia intermedia (hemoglobin H disease) and β-thalassemia intermedia (β-TI), hemoglobin E/β- thalassemia (mild and moderate forms). Differentiating a new patient with thalassemia as TDT or NTDT is essential and requires an accurate clinician's evaluation using various indicators such as hematological parameters, particularly baseline Hb levels, and follow-up for a minimum of 3 to 6 months to determine clinical severity is recommended before making a diagnosis of TDT or NTDT ( 5 , 6 ).
The three important pathophysiologic factors in β-thalassemias are: chronic anemia/hypoxia, ineffective erythropoiesis, and iron overload. The harshness of the disease depends mainly on molecular deficiencies. Chain imbalance causes excess unstable α chains to provoke within erythroid progenitors, leading to cell membrane decline and cell lysis. This triggers an alteration in the mycomedial environment of the bone marrow due to an imbalance of cytokines that causes the erythroid progenitors to proliferate but with inadequate maturation, which is called ineffective erythropoiesis. This cytokine imbalance together with bone marrow hyperplasia causes extramedullary erythropoiesis and subsequently the associate bone deformations. Because of anemia/chronic hypoxia, infective erythropoiesis retroelements, is maintained and perpetuates over time ( 3 , 6 ).
Ineffective Erythropoiesis
Erythropoiesis is a finely regulated process in which every stage is highly regulated by different signal transduction pathways and proteins. Erythropoiesis process in humans is divided into two parts: the early stage of erythropoiesis and the late stage. The first stage is EPO-dependent whereas the second stage is iron-dependent. Erythropoietin (EPO-dependent stage) is the main regulator of early-stage erythropoiesis whereas erythrocyte differentiation and maturation are negatively regulated by the transforming growth factor (TGF-family), the late-stage erythropoiesis or iron-dependent stage of erythropoiesis ( Figure 2 ).

Figure 2 . Schematic representation of erythropoiesis. During erythroid development, several stages occur in which a complex network of molecules are expressed (EPO, iron, transcription factors) are involved. In early-stage erythropoiesis, EPO is the main regulator after BFU-E formation. In this stage, GATA-1 promotes erythropoiesis and increases the EPO receptor expression. In large-stage erythropoiesis molecules such as transferrin and growth/differentiating factor 11 (GDF11) are involved. Erythroid expansion is negatively regulated by association of FAS to FAS ligand, which has as a consequence the apoptosis on immature erythroid cells, and GDF11 and other members of the TGF-β family, which negatively regulate erythrocyte differentiation and maturation from the early to the late stages.
EPO-Dependent Erythropoiesis
In this stage, EPO is fundamental for the proliferation of erythroid progenitors. The recognition of EPO by EPO-R on the surface of these precursors induces JAK2 activation activating STAT5 phosphorylation, with associated induction of erythroid antiapoptotic genes and increase of erythroid progenitors proliferation and survival ( 7 ). The production of EPO and the expansion of erythropoiesis is regulated according to demand (hypoxia, hemorrhage, hemolysis). On the contrary, if the production of erythrocytes is adequate, down-regulation by a mechanism of apoptosis is performed. The predisposition of EPO-dependent erythroid progenitors to apoptosis is associated with different levels of FAS protein (CD95) expression and the FAS ligand (FASL), which belong to the family of TNF receptors, the binding of which activates the caspase cascade and consequently, the immature cell apoptosis limiting erythropoiesis expansion. On the other hand, if the need for erythrocytes increases, EPO production increases, and apoptosis is reduced because EPO stimulates the production of heat shock protein 70 (HSP70) that protects GATA1 from cleavage by FAS/FASL and the activation of the caspase cascade ( 8 ).
Fe-Dependent Erythropoiesis
In the large stage, the presence of iron is essential for the synthesis of hemoglobin, and the integrity of the erythroferrone (ERFE)-hepcidin-ferroportin axis is essential for iron homeostasis ( 9 , 10 ). ERFE is a potent negative regulator of hepcidin. When it is chronically elevated, such as in situations of ineffective erythropoiesis, low plasma iron availability occurs. Transferrin (and its cellular receptor) is also involved in this stage as well as growth differentiation factors like GDF11, a member of the TGF-β superfamily, which negatively regulate erythrocyte maturation and differentiation ( 11 ).
TGF-β receptor ligands are a group of cytokines that include TGF, activins, bone morphogenetic proteins (BMPs), and GDF-11 and play an important role in the regulation of erythropoiesis within the hematopoietic stem cell niche. Various activins, and in particular GDF-11, exert inhibitory activity at the late stage of erythropoiesis.
In the TGF signaling pathway, ligand binding to the type II receptor leads to the recruitment and phosphorylation of the type I receptor and phosphorylation of regulatory SMADs (R-SMADs), SMAD2 and 3, to form the R-SMAD/SMAD4 complex, which modulates the expression of target genes inducing an inhibitory activity on erythroid differentiation by inducing apoptosis in erythroblasts.
During normal erythroid maturation, reduced GDF11 expression with consequent TGF- β signaling suppression, and EPO stimulation occur in parallel, and both are essential for the differentiation of hematopoietic erythroid progenitor cells ( 12 ).
In β-thalassemia, ineffective erythropoiesis is triggered by two main pathogenic mechanisms ( 7 ). On the one hand, α-globin chains aggregates sequester cytosolic heat shock protein 70 (HSP70). This inhibits its nuclear translocation and protects the erythroid transcription factor GATA-factor 1 (GATA1) from cleavage. On the other hand, these toxic aggregates of α-globin chains stimulate the formation of radical oxygen species (ROS) (whose formation is also produced by other mechanisms such as iron overload), which activate GDF11, which in turn activates the inhibitory pathway of SMAD2/3, and as a consequence, erythroid differentiation is inhibited ( 13 ).
Iron Overload
Iron overload is one of the main pathogenic events in β-thalassemia. Apart from the transfusion-dependent iron overload in patients with TDT, there is a mechanism by which an inappropriate increase in intestinal iron absorption occurs, both in patients with TDT and in NTDT.
This mechanism is triggered by ineffective erythropoiesis. The accumulation of erythroid precursors during ineffective erythropoiesis increases the production of erythropherrone, potent negative regulator of hepcidin secreted by bone marrow erythroblasts ( 10 ), which negatively regulates the expression of hepcidin. The decrease in hepcidin, the main negative ferroportin modulator (an iron transporter protein in the basolateral membrane of the enterocyte), increases iron absorption (hepcidin inhibits iron absorption and recycling ferroportin) and release of iron from the reticuloendothelial in situations of iron overload and iron sequestration in erythropoiesis. It has been described that iron availability during stress erythropoiesis is produced by increase of ERFE ( 13 ).
In NTDT, growth differentiation factor 15 (GDF-15) can induce hepcidin downregulation. This is a member of the transforming growth factor- (TGF-β) family which use to be upregulated during ineffective erythropoiesis, causing the down regulation of hepcidin ( 14 ).
Bone Disease in β-Thalassemia
Recently, several advances in the understanding of the pathophysiology of bone disease in β-thalassemia have been done. Classically, it was directly attributed to ineffective erythropoiesis and secondary bone expansion, but recently it has been shown that there is an imbalance of cytokines that can directly alter bone metabolism, although the mechanisms involved are not yet well-established.
Both patients with TDT and NTDT show marked decreases in bone mineral density (BMD), despite optimization of transfusions, and low BMD continues to be a frequent complication in these patients.
The mechanisms that have been postulated to explain the loss of bone mineral density in patients with β-thalassemia include explicit effects of abnormal erythroid proliferation with bone expansion, increased circulating erythropoietin (EPO), iron bone deposit with iron toxicity, and oxidative stress with endocrine secondary disorders (hypogonadism, deficit GH-IGF-1, vitamin D deficiency) that which in turn affect the bone mineral loss and secondary osteoporosis ( 15 ).
In recent years, there is growing evidence of the relationship between erythropoiesis, bone mineral metabolism and iron homeostasis. Recently, a mechanism responsible for the activation of osteoclasts in thalassemic patients has been described that could be associated to cytokine dysregulation and, in particular, to the modification of the RANK/RANKL/OPG axis, which is essential for the regulation of osteoclastogenesis ( 16 , 17 ). The OPG/RANK/RANKL system is essential for the regulation of osteoclastogenesis. Osteoprotegerin (OPG) or osteoclastogenesis inhibition factor (OCIF or TNFRSF11B), is a member of the superfamily of tumor necrosis factor (TNFR) receptors that is expressed and secreted in numerous tissues (lung, heart, kidneys, liver, intestine, stomach, brain, thyroid gland and spinal cord) as well as in bone in which its main function is to inhibit the maturation and activation of osteoclasts.
Recently, it has been shown ERFE binds and sequesters some members of the bone morphogenetic protein (BMP) family, primarily BMP2, BMP6, and the BMP2/6 what suppresses hepcidin by inhibiting hepatic BMP/SMAD signaling. Therefore, iron availability by stimulated erythropoiesis can be regulated by ERFE ( 9 ). Bone formation by osteoblasts during skeletal development, modeling, and ongoing remodeling can be stimulated by BMPs. Therefore, ERFE seems to be key in the recently described erythropoiesis-iron-bone circuit, by modifying the availability of BMP. Therefore, ERFE appears to be an important link between abnormal erythropoiesis, iron metabolism alteration, and loss of BMD in β-thalassemia ( 16 , 17 ).
The mechanism of action of ERFE, through the sequestration of BMP, could be that the loss of ERFE, by improving the availability of BMP, stimulates the formation of osteoblastic bone ( 18 ). However, high ERFE levels are osteoprotective and prevent bone loss in β-thalassemia when erythropoiesis is extended. Therefore, there is a paradoxical effect that has not yet been fully explained. Although in TDT patients, in whom ERFE is inhibited post-transfusion, and is lower than in NTDT patients, could explain the more severe bone alterations in these patients despite transfusions and ERFE has a protective function decreasing bone loss phenotype in b-thalassemia.
In addition, other data indicate that the loss of BMP signaling (high ERFE), increases bone mass through direct inhibition of osteoclasts and activation of the Wnt pathway, predicting that the loss of ERFE would lead to a decrease in bone mass ( 16 , 17 ) by increased expression of RANKL and sclerostin.
In summary, to date the only known function of ERFE was hepcidin regulation expression through BMP sequestration, contributing to iron overload ( 9 ). However, ERFE seems to have bone metabolisms implications and a new role in bone protection has been described ( 16 ). In conditions of elevated ERFE, such as β-thalassemia and others ineffective erythropoiesis situations, BMP2 and BMP6 proteins are sequestered, decreasing signaling through the BMP/Smad and ERK pathways. This would result in decreased SOST and RANKL expression (Rankl/OPG) with decrease osteoclastogenesis and bone resorption. On the other hand, when level ERFE is low, increased BMP2, and BMP6 proteins, lead to stimulate osteoclastogenesis (RANKL/Opg), bone resorption and increased sclerostin osteocytes synthesis (expression SOST gene), with a consequent decrease in bone formation by inhibition of osteoblastic function.
GATA1 Levels Regulation in β-Thalassemia
As mentioned above, erythropoiesis starts from hematopoietic stem cells (HPSCs), in a finely regulated process, that involves various factors, and it is controlled at different molecular levels by growth factors and hormones such as erythropoietin, that activate different signaling pathways that end up activating erythroid transcription factors. The essential transcription factors for erythropoiesis are GATA-1, SCL, TAL1, LMO2, LDB1, KLF-1, and GFI-1B, although many more participate. These factors are organized in a complex called CEN (“Core ErythroidNetwork”) ( 19 ), whose operation is finely regulated by SCF and EPO between others, to ensure adequate erythrocyte development. GATA1 is considered the “master regulator” of this process, GATA1 is a DNA-binding zinc finger transcription factor that plays an essential role in the normal development of hematopoietic lines. The protein contains 413 amino acids, with an N-terminal region where its transcriptional activity resides and a C-terminal region that mediates the binding of GATA1 to DNA and other proteins. In 1995, two isoforms of GATA1 resulting from alternative splicing were identified. GATA1 encodes a 47 kDa protein and GATA1s, a shorter 40 kDa protein that lacks the transactivation domain at the N-terminus. Both proteins are capable of binding to DNA and could form dimers or heterodimers, although the shorter GATA1s isoform is less active than the long ( 20 ). In 1999 cleavage of GATA1 by Caspase 3 was reported ( 21 ). More recently, it has been linked the stabilization of GATA1 to inhibition of Caspase-1 Activity ( 22 ).
β-thalassemia shares several common elements with other forms of anemia including a deficiency of GATA1 the ‘master regulator' in erythropoiesis ( 23 ), GATA1 levels are finely regulated during erythropoiesis to develop sufficient functional erythrocytes ( 24 ). This transcription factor is necessary for normal early erythroid progenitors' differentiation [i.e., colony-forming unit-erythroid (CFU-E) and burst-forming unit erythroid (BFU-E) cells]. In β-thalassemia, α-hemoglobin chains accumulate in the cytosol due to the non-functionality of β-hemoglobin chains and sequester heat shock protein 70 (HSP70). Subsequently, HSP70 cannot be translocated into the nucleus thereby impairing GATA1 stabilization through caspase 3 cleavage, resulting in altered GATA1 levels with disturbed erythropoiesis and accumulation of unfunctional erythroid progenitors ( 23 ).
Importantly, GATA1 is highly expressed during these early stages of differentiation but GATA1 protein expression is shown to decline toward terminal erythroid differentiation ( 25 ). Recent data from Tyrkalska et al. demonstrated the role of the inflammasome, a complex of innate immune system which receptors and sensors playing important roles in infection and inflammation, in GATA1 regulation, and subsequent erythroid differentiation. Pharmacological inhibition of the inflammasome was shown to stimulate GATA1 expression and promote erythroid differentiation ( 22 ).
On the other hand, Fetal hemoglobin (HbF) increase has revealed as a promising results to treat β-hemoglobinopathies ( 26 ) with a recent implication of MiR-486-3p and miR-15a in Fetal hemoglobin induction ( 27 ). Several groups reported that increase of miR-210 levels is high in erythroid precursors from β-thalassemia patients what have an impact on fetal hemoglobin (HbF) levels ( 28 – 31 ).
Finally, other genetic studies indicate a relationship between variants in the gene BCL11A and HbF levels ( 29 ). There they described that reduced BCL11A expression induce HbF. The data published by Gasparello et al. are consistent with a globin gene regulation by BCL11A and therefore postulated BCL11A as a therapeutic candidate to reactivate HbF in disorders associated to beta-hemoglobin ( 29 ). Aligned with this, Bauer et al. published that GWAS-marked BCL11A enhancer represents another potential optional treatment in this disease ( 32 ).
All the processes described above are reflected and connected in Figure 3 .

Figure 3 . Clinical complications and pathophysiological mechanisms of β-Thalassemia. In thalassemia, the imbalance α/β-globin synthesis is the fundamental initial pathogenic event. Excess α-globin chains precipitate in the cytoplasm, sequester HSP70 and GATA1 is cleaved by Caspase 3/1 which result in dysfunctional erythropoiesis and imposes metabolic stress on the erythrocytes, specifically in the form of excess generation of reactive oxygen species and increased demand on adenosine triphosphate (ATP)-dependent proteolytic mechanisms to clear excess globin chains. These pathophysiological changes lead to the characteristics of this disease: ineffective erythropoiesis, peripheral hemolysis, and subsequent anemia. Clinical implications of the α- and β-globin imbalance include lack of sufficient RBCs and Hb for effective oxygen transport, and ineffective erythropoiesis and hemolysis, which can lead to splenomegaly, bone marrow expansion (extramedullary hematopoiesis), concomitant bone deformities, and iron overload.
Discussion: Perspectives and Future New Potential β-Thalassemia Treatments
Despite the high social and economic impact of β-thalassemia, there is no curative treatment available, except for bone marrow transplant for the few pediatric patients who have an identical HLA donor, and who assume the high morbidity and mortality of transplant that is sometimes not acceptable for non-malignant disease. Currently no approved treatments to handle anemia in NTDT and, even though it has recently been approved, the first drug that improves anemia in these patients (luspatercept), has limited efficacy. Therefore, there are no effective treatments to improve anemia or to reduce red blood cell transfusions and chronic complications. However, in recent years there is a growing interest in studying these diseases with an increasing number of clinical trials directed against various therapeutic targets (gene therapy, erythroid maturation agents, pyruvate kinase activators, JAK kinase 2 inhibitors, targeting iron dysregulation). All of them are being developed and in the future may change the quality of life of these patients and we aim to be part of this scenario. All of them summarized in a succinct way by Musallam et al. ( 33 ).
Although these targets may be effective, efficacy is partial, not all patients respond, and therefore further research and treatments are required considering its multifactorial pathophysiology. Perhaps the correct approach in the future is combined treatment and, in this scenario, it will be essential to explore new therapeutic targets. In recent years, inflammasomes have emerged as a new potential therapeutic target for these types of diseases as well as the HSP70 nuclei regulation.
Inflammasome as a Potential Target Treatment in β-Thalassemia
Inflammasomes are multiprotein complexes firstly described a decade ago by Tschopp and colleagues. Inflammasomes are multiprotein complexes usually composed of sensor proteins, mainly from the NLR family, adaptor proteins such as ASC, and an effector cysteine-protease enzyme, usually caspase-1 ( 34 ). Gasdermin D (GSDMD) a pore-forming protein is cleavage and activated by caspase-1 ( 35 ), what induce cytokine release and pyroptosis ( 36 ). Several inflammasome receptors have been described with different roles including inflammatory diseases, sepsis protection or host defense ( 37 – 39 ).
Inflammasomes are broadly expressed in hematopoietic and non-hematopoietic cells and can induce different responses including production of IL-18, IL-1β, eicosanoids, and pyroptosis. Since this first description, research within the inflammasome field has been one of the most studied fields in immunology leading to huge advances.
In the innate immune context, the activation of inflammasomes is essential in the clearance of pathogens or damaged cells. On the other hand, uncontrolled inflammasome activation induces metabolic and autoimmune disorders, indicating the importance of these complexes. The recent role of the inflammasome in erythropoiesis brings the focus to these complexes to treat anemia and neutrophilia ( 22 ).
The effector protein of the inflammasomes are caspases and previous data indicated a role of Caspase 3 in GATA1 stabilization ( 21 ). More recently, inflammasome has been postulated as a potential treatment of Diamond-Blackfan anemia, this type of anemia curses by a deficiency in GATA1 levels due to a ribosomopathy that produces an inefficient translation of GATA1 ( 22 , 40 ). Tyrskalska et al. ( 22 )demonstrated an increase of GATA1 levels in human and zebrafish larvae with caspase-1 inhibitor. Therefore, potentially inhibition of Caspase-1 or Caspase-3 could be considered as a potential treatment to stabilize GATA1 levels to increase red blood cells formation. In this scenario, the identification of the inflammasome type that mediates this process will be essential to translate these results with higher specificity to the clinic ( Figure 4 ).

Figure 4 . New potential treatments for β-thalassemia. In normal erythroblast, GATA1 levels are regulated through the balance Caspase 3/1 cleavage and HSP70 protecting function in the nucleus. As a consequence, normal and functional erythrocytes are produced. In contrast, in β-thalassemic erythroblast, the lack of functional β-globin chains induces accumulation of free α-globin chains which restrict HSP70 distribution to the cytoplasm and therefore GATA1 is cleaved by Caspase 3/1 which result in fewer functional matured erythrocytes.
Inhibition of HSP70 Export From the Nucleus
As described above, GATA1 levels are essential in erythroid differentiation, and in this plays an important role the Heat Shock Protein 70 (HSP70) a chaperone, which is translocated to the nucleus to protect GATA1 transcription factor of caspase-3 cleavage ( 41 ). In β-thalassemia, the accumulation of free α-globin chains sequestered HSP70 in the cytosol which avoids the protective role of GATA1 into the nucleus. A recent publication demonstrated that HSP70 localization is regulated by the exportin-1 (XPO1) and inhibition of XPO1 increase HSP70 levels in normal erythroid progenitors what increase, which have as a consequence an increase in GATA1 levels ( 42 ). This introducesXPO1 inhibitors as a new therapeutic option to treat β-thalassemia ( 42 , 43 ).
For many years, management of β-Thalassemia patients has been limited to blood transfusion and iron chelation. However, β-Thalassemia is now the focus of a flourishing research field that has already offered new treatments with the potential to modify the natural history of the disease and the quality of life of the patients. In this review we have summarized the present knowledge of the pathophysiology of the disease and proposed future possible new, and more directed approaches, to its treatment ( Figure 4 ).
Conclusions
β-thalassemia (transfusion and non-transfusion dependent), is an inherited hemoglobinopathy caused by a quantitative defect in the synthesis of β-globin chains of hemoglobin, leading to the accumulation of free α-globin chains aggregates that cause ineffective erythropoiesis. The only curative treatment for these patients is hematopoietic stem cell transplantation, but this option only is feasible in a few patients with HLA-matched sibling donors. In most patients, the development of chelation and support treatments has improved survival, however, chronic complications have increased (iron overload, osteoporosis, extramedullary hematopoiesis, etc.) that limit their quality of life.
Despite increasing knowledge of the pathophysiology of β-thalassemia, only luspatercept has been recently approved in TDT patients, reducing transfusion needs but of limited effectiveness. NTDT patients, which do not require regular transfusions, lack an approved treatment and have the same (if not more) complications as TDT patients. Therefore, new treatments are needed that, alone or in combination with existing ones, can improve the expectations of these patients.
In human erythroblast, terminal erythroid maturation is altered due to HSP70 sequestration in the cytoplasm by free α-globin chains preventing its accumulation in the nucleus to protect GATA1 transcription factor from Caspase-3 cleavage, resulting in maturation arrest and apoptosis. ERFE and BMP play an important role in bone disease but are not well-established yet.
The knowledge of these critical new pathophysiological approaches can help develop new therapeutic options such as XPO1 inhibitors or inflammasome inhibitors that could rescue GATA-1 expression, improved erythroid terminal differentiation, and represent a new therapeutic option to ameliorate ineffective erythropoiesis, iron overload, and decreased mineral bone mass of β-thalassemia patients.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Author Contributions
MS-V: writing—original draft preparation. AP-O, ES, JM, and MB: writing—review and editing. All authors have read and agreed to the published version of the manuscript.
This work was supported by the Institute of Health Carlos III (ISCIII) through a Miguel Servet Contract Program and the Associated Budget CP20/00028 to AP-O partially funded by European Funding. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Origa R. beta-Thalassemia. Genet Med. (2017) 19:609–19. doi: 10.1038/gim.2016.173
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Kattamis A, Forni GL, Aydinok Y, Viprakasit V. Changing patterns in the epidemiology of beta-thalassemia. Eur J Haematol. (2020) 105:692–703. doi: 10.1111/ejh.13512
3. Asadov C, Alimirzoeva Z, Mammadova T, Aliyeva G, Gafarova S, Mammadov J. beta-Thalassemia intermedia: a comprehensive overview and novel approaches. Int J Hematol. (2018) 108:5–21. doi: 10.1007/s12185-018-2411-9
4. Viprakasit V, Ekwattanakit S. Clinical classification, screening and diagnosis for thalassemia. Hematol Oncol Clin North Am. (2018) 32:193–211. doi: 10.1016/j.hoc.2017.11.006
5. Cappellini MD, Cohen A, Porter J, Taher A, Viprakasit V,. (eds.). Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT) . Nicosia: Thalassaemia International Federation (2014).
Google Scholar
6. Taher A, Vichinsky E, Musallam K, Cappellini MD, Viprakasit V. Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT) . Nicosia: Thalassaemia International Federation (2013).
PubMed Abstract | Google Scholar
7. Elliott S, Pham E, Macdougall IC. Erythropoietins: a common mechanism of action. Exp Hematol. (2008) 36:1573–84. doi: 10.1016/j.exphem.2008.08.003
8. Valent P, Busche G, Theurl I, Uras IZ, Germing U, Stauder R, et al. Normal and pathological erythropoiesis in adults: from gene regulation to targeted treatment concepts. Haematologica. (2018) 103:1593–603. doi: 10.3324/haematol.2018.192518
9. Arezes J, Foy N, McHugh K, Sawant A, Quinkert D, Terraube V, et al. Erythroferrone inhibits the induction of hepcidin by BMP6. Blood. (2018) 132:1473–7. doi: 10.1182/blood-2018-06-857995
10. Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. (2014) 46:678–84. doi: 10.1038/ng.2996
11. Blank U, Karlsson S. TGF-beta signaling in the control of hematopoietic stem cells. Blood. (2015) 125:3542–50. doi: 10.1182/blood-2014-12-618090
12. Xie Y, Bai H, Liu Y, Hoyle DL, Cheng T, Wang ZZ. Cooperative effect of erythropoietin and TGF-beta inhibition on erythroid development in human pluripotent stem cells. J Cell Biochem. (2015) 116:2735–43. doi: 10.1002/jcb.25233
13. Camaschella C, Nai A. Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. Br J Haematol. (2016) 172:512–23. doi: 10.1111/bjh.13820
14. Tanno T, Bhanu NV, Oneal PA, Goh SH, Staker P, Lee YT, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. (2007) 13:1096–101. doi: 10.1038/nm1629
15. Gaudio A, Xourafa A, Rapisarda R, Zanoli L, Signorelli SS, Castellino P. Hematological diseases and osteoporosis. Int J Mol Sci. (2020) 21:3538. doi: 10.3390/ijms21103538
16. Castro-Mollo M, Gera S, Ruiz-Martinez M, Feola M, Gumerova A, Planoutene M, et al. The hepcidin regulator erythroferrone is a new member of the erythropoiesis-iron-bone circuitry. Elife. (2021) 10:e68217. doi: 10.7554/eLife.68217
17. Watkins LR, Wiertelak EP, Goehler LE, Mooney-Heiberger K, Martinez J, Furness L, et al. Neurocircuitry of illness-induced hyperalgesia. Brain Res. (1994) 639:283–99. doi: 10.1016/0006-8993(94)91742-6
18. Salazar VS, Gamer LW, Rosen V. BMP signalling in skeletal development, disease and repair. Nat Rev Endocrinol. (2016) 12:203–21. doi: 10.1038/nrendo.2016.12
19. Wontakal SN, Guo X, Smith C, MacCarthy T, Bresnick EH, Bergman A, et al. A core erythroid transcriptional network is repressed by a master regulator of myelo-lymphoid differentiation. Proc Natl Acad Sci U S A. (2012) 109:3832–7. doi: 10.1073/pnas.1121019109
20. Calligaris R, Bottardi S, Cogoi S, Apezteguia I, Santoro C. Alternative translation initiation site usage results in two functionally distinct forms of the GATA-1 transcription factor. Proc Natl Acad Sci U S A. (1995) 92:11598–602. doi: 10.1073/pnas.92.25.11598
21. De Maria R, Zeuner A, Eramo A, Domenichelli C, Bonci D, Grignani F, et al. Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature. (1999) 401:489–93. doi: 10.1038/46809
22. Tyrkalska SD, Perez-Oliva AB, Rodriguez-Ruiz L, Martinez-Morcillo FJ, Alcaraz-Perez F, Martinez-Navarro FJ, et al. Inflammasome regulates hematopoiesis through cleavage of the master erythroid transcription factor GATA1. Immunity. (2019) 51:50–63.e55. doi: 10.1016/j.immuni.2019.05.005
23. Arlet JB, Ribeil JA, Guillem F, Negre O, Hazoume A, Marcion G, et al. HSP70 sequestration by free alpha-globin promotes ineffective erythropoiesis in beta-thalassaemia. Nature. (2014) 514:242–6. doi: 10.1038/nature13614
24. Ferreira R, Ohneda K, Yamamoto M, Philipsen S. GATA1 function, a paradigm for transcription factors in hematopoiesis. Mol Cell Biol. (2005) 25:1215–27. doi: 10.1128/MCB.25.4.1215-1227.2005
25. Gutierrez L, Caballero N, Fernandez-Calleja L, Karkoulia E, Strouboulis J. Regulation of GATA1 levels in erythropoiesis. IUBMB Life. (2020) 72:89–105. doi: 10.1002/iub.2192
26. Salinas Cisneros G, Thein SL. Research in sickle cell disease: from bedside to bench to bedside. Hemasphere. (2021) 5:e584. doi: 10.1097/HS9.0000000000000584
27. Eltaweel NH, ElKamah GY, Khairat R, Atia HAE, Amr KS. Epigenetic effects toward new insights as potential therapeutic target in B-thalassemia. J Genet Eng Biotechnol. (2021) 19:51. doi: 10.1186/s43141-021-00138-x
28. Bavelloni A, Poli A, Fiume R, Blalock W, Matteucci A, Ramazzotti G, et al. PLC-beta 1 regulates the expression of miR-210 during mithramycin-mediated erythroid differentiation in K562 cells. Oncotarget. (2014) 5:4222–31. doi: 10.18632/oncotarget.1972
29. Gasparello J, Fabbri E, Bianchi N, Breveglieri G, Zuccato C, Borgatti M, et al. BCL11A mRNA targeting by miR-210: A possible network regulating γ-globin gene expression. Int J Mol Sci . (2017) 18:2530. doi: 10.3390/ijms18122530
30. Sarakul O, Vattanaviboon P, Tanaka Y, Fucharoen S, Abe Y, Svasti S, et al. Enhanced erythroid cell differentiation in hypoxic condition is in part contributed by miR-210. Blood Cells Mol Dis. (2013) 51:98–103. doi: 10.1016/j.bcmd.2013.03.005
31. Sawant M, Chandrakala S, Colah R, Ghosh K, Nadkarni A. Does HbF induction by hydroxycarbamide work through MIR210 in sickle cell anaemia patients? Br J Haematol. (2016) 173:801–3. doi: 10.1111/bjh.13642
32. Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. (2013) 342:253–7. doi: 10.1126/science.1242088
33. Musallam KM, Bou-Fakhredin R, Cappellini MD, Taher AT. 2021 update on clinical trials in beta-thalassemia. Am J Hematol. (2021) 96:1518–31. doi: 10.1002/ajh.26316
34. Schroder K, Tschopp J. The inflammasomes. Cell. (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
35. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
36. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
37. Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. (2004) 20:319–25. doi: 10.1016/S1074-7613(04)00046-9
38. Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. (2005) 73:1907–16. doi: 10.1128/IAI.73.4.1907-1916.2005
39. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. (2004) 430:213–8. doi: 10.1038/nature02664
40. Danilova N, Gazda HT. Ribosomopathies: how a common root can cause a tree of pathologies. Dis Model Mech. (2015) 8:1013–26. doi: 10.1242/dmm.020529
41. Ribeil JA, Zermati Y, Vandekerckhove J, Cathelin S, Kersual J, Dussiot M, et al. Hsp70 regulates erythropoiesis by preventing caspase-3-mediated cleavage of GATA-1. Nature. (2007) 445:102–5. doi: 10.1038/nature05378
42. Guillem F, Dussiot M, Colin E, Suriyun T, Arlet JB, Goudin N, et al. XPO1 regulates erythroid differentiation and is a new target for the treatment of beta-thalassemia. Haematologica. (2020) 105:2240–9. doi: 10.3324/haematol.2018.210054
43. Modepalli S, Hattangadi SM. Novel use for selective inhibitors of nuclear export in beta-thalassemia: block of HSP70 export from the nucleus via exportin Xpo1 improves ineffective erythropoiesis. Haematologica. (2020) 105:2188–9. doi: 10.3324/haematol.2020.254474
Keywords: anemia, thalassemia, β-globin, GATA1, inflammasome
Citation: Sanchez-Villalobos M, Blanquer M, Moraleda JM, Salido EJ and Perez-Oliva AB (2022) New Insights Into Pathophysiology of β-Thalassemia. Front. Med. 9:880752. doi: 10.3389/fmed.2022.880752
Received: 21 February 2022; Accepted: 22 March 2022; Published: 12 April 2022.
Reviewed by:
Copyright © 2022 Sanchez-Villalobos, Blanquer, Moraleda, Salido and Perez-Oliva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY) . The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eduardo J. Salido, eduardoj.salido@carm.es ; Ana B. Perez-Oliva, anab.perez@imib.es
This article is part of the Research Topic
Constructing New Motifs in Hematology
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Correspondence
- Published: 16 September 2022
Long-term health-related quality of life in patients with β-thalassemia after unrelated hematopoietic stem cell transplantation
- Olga Mulas ORCID: orcid.org/0000-0002-7487-3328 1 , 2 na1 ,
- Giovanni Caocci ORCID: orcid.org/0000-0002-6585-5187 1 , 2 na1 ,
- Fabio Efficace 3 ,
- Eugenia Piras 2 ,
- Clara Targhetta 2 ,
- Veronica Frau 2 ,
- Susanna Barella 4 ,
- Antonio Piroddi 5 ,
- Maria Grazia Orofino 5 ,
- Adriana Vacca 2 &
- Giorgio La Nasa 1 , 2
Bone Marrow Transplantation volume 57 , pages 1833–1836 ( 2022 ) Cite this article
186 Accesses
3 Citations
1 Altmetric
Metrics details
- Haematological diseases
- Quality of life
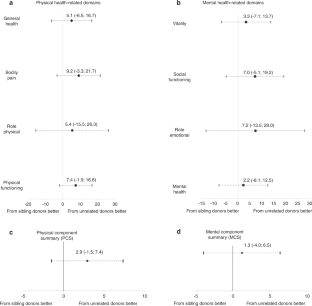
In western nations, regular iron chelation therapy and optimization of erythrocyte transfusion support have improved the life expectancy of patients with β-Thalassemia Major (β-TM) born in the last few decades, approaching that of healthy peers [ 1 ]. Nevertheless, the curative role of hematopoietic stem cell transplantation (HSCT) in β-TM still remains very relevant [ 2 ]. An international consensus recommends that HSCT be offered as soon as possible before developing iron overload and iron-related tissue damage in patients with an available HLA identical sibling donor [ 3 ]. However, less than 30% of patients can find a sibling donor within the family, depending on nationality and ethnic group. Lacking a sibling donor, a matched unrelated HSCT represents a valid alternative [ 4 , 5 ]. However, this procedure is an option to be performed in centers with appropriate expertise [ 3 , 4 ]. Among different parameters used to evaluate the success of the transplant, health-related quality of life (HRQoL) plays a key role. Very little data is available on HRQoL of sibling HSCT and, to our best knowledge, only one study reported HRQoL outcomes in β-TM patients transplanted from unrelated donors [ 6 ]. Moreover, no direct comparisons on HRQoL are available between patients who underwent siblings or matched unrelated HSCT.
The main objective of our study was to analyze long-term HRQoL in matched unrelated HSCT in β-TM carried out in centers with high expertise in the procedure. The secondary objective was to compare HRQoL between unrelated and sibling HSCT.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
251,40 € per year
only 20,95 € per issue
Rent or buy this article
Prices vary by article type
Prices may be subject to local taxes which are calculated during checkout
Data availability
The data that support the findings of this study are available from the corresponding author, GC, upon reasonable request
Taher AT, Musallam KM, Cappellini MD. β-Thalassemias. N Engl J Med. 2021;384:727–43.
Mulas O, Mola B, Caocci G, La, Nasa G. Conditioning regimens in patients with β-thalassemia who underwent hematopoietic stem cell transplantation: a scoping review. J Clin Med. 2022;11:907.
Article CAS PubMed PubMed Central Google Scholar
Angelucci E, Matthes-Martin S, Baronciani D, Bernaudin F, Bonanomi S, Cappellini MD, et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica. 2014;99:811–20.
Article PubMed PubMed Central Google Scholar
Shenoy S, Angelucci E, Arnold SD, Baker KS, Bhatia M, Bresters D, et al. Current results and future research priorities in late effects after hematopoietic stem cell transplantation for children with sickle cell disease and thalassemia: a consensus statement from the second pediatric blood and marrow transplant consortium international conference on late effects after pediatric hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2017;23:552–61.
Article PubMed Google Scholar
Caocci G, Orofino MG, Vacca A, Piroddi A, Piras E, Addari MC, et al. Long-term survival of beta thalassemia major patients treated with hematopoietic stem cell transplantation compared with survival with conventional treatment. Am J Hematol. 2017;92:1303–10.
Article CAS PubMed Google Scholar
Caocci G, Pisu S, Argiolu F, Giardini C, Locatelli F, Vacca A, et al. Decision-making in adult thalassemia patients undergoing unrelated bone marrow transplantation: quality of life, communication and ethical issues. Bone Marrow Transplant. 2006;37:165–9.
Bahar B, Gholibeikian S, Aghdami N, Heshmat R, Safavifar F, Samiee S. Quality of life in beta thalassaemia major at least six months post-allogeneic stem cell transplantation in comparison to the same non-transplanted patients. Bone Marrow Transplant. 2005;35:S268.
Google Scholar
Caocci G, Efficace F, Ciotti F, Roncarolo MG, Vacca A, Piras E, et al. Prospective assessment of health-related quality of life in pediatric patients with beta-thalassemia following hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2011;17:861–6.
Caocci G, Vacca A, Piras E, Serreli V, Dessi C, Marcias M, et al. Return to normal life after hematopoietic stem cell transplantation for thalassemia: a study of patients transplanted from matched sibling donors. Bone Marrow Transplant. 2016;51:1640–1.
Cheuk DKL, Mok ASP, Lee ACW, Chiang AKS, Ha SY, Lau YL, et al. Quality of life in patients with transfusion-dependent thalassemia after hematopoietic SCT. Bone Marrow Transplant. 2008;42:319–27.
Kelly MJ, Pennarola BW, Rodday AM, Parsons SK. on behalf of the Journeys to Recovery Study, the HSCT-CHESS TM Study. Health-related quality of life (HRQL) in children with sickle cell disease and thalassemia following hematopoietic stem cell transplant (HSCT). Pediatr Blood Cancer. 2012;59:725–31.
La Nasa G, Caocci G, Efficace F, Dessì C, Vacca A, Piras E, et al. Long-term health-related quality of life evaluated more than 20 years after hematopoietic stem cell transplantation for thalassemia. Blood. 2013;122:2262–70.
Uygun V, Tayfun F, Akcan M, Karasu GT, Kupesiz A, Hazar V, et al. Quality of life assessment in hematopoietic stem cell transplantation performed on thalassemia major patients. Pediatr Hematol Oncol. 2012;29:461–71.
Patel S, Swaminathan VV, Mythili V, Venkatadesikalu M, Sivasankaran M, Jayaraman D, et al. Quality matters—hematopoietic stem cell transplantation versus transfusion and chelation in thalassemia major. Indian Pediatr. 2018;55:1056–8.
Javanbakht M, Keshtkaran A, Shabaninejad H, Karami H, Zakerinia M, Delavari S. Comparison of blood transfusion plus chelation therapy and bone marrow transplantation in patients with beta-thalassemia: application of SF-36, EQ-5D, and visual analogue scale measures. Int J Health Policy Manag. 2015;4:733–40.
Download references
Acknowledgements
We thank Dr. Emanuela Morelli and Dr. Luigi Marras for collecting web data and supporting the analysis of data
Author information
These authors contributed equally: Olga Mulas, Giovanni Caocci
Authors and Affiliations
Department of Medical Sciences and Public Health, University of Cagliari, Cagliari, Italy
Olga Mulas, Giovanni Caocci & Giorgio La Nasa
Hematology Unit, Businco Hospital, Cagliari, Italy
Olga Mulas, Giovanni Caocci, Eugenia Piras, Clara Targhetta, Veronica Frau, Adriana Vacca & Giorgio La Nasa
Health Outcomes Research Unit, Italian Group for Adult Hematologic Diseases Data Center, Rome, Italy
Fabio Efficace
Pediatric Clinic, Thalassemia and Rare Diseases, Pediatric Hospital “Microcitemico A. Cao”, Cagliari, Italy
Susanna Barella
Bone Marrow Transplant Center, Pediatric Hospital “Microcitemico A. Cao”, Cagliari, Italy
Antonio Piroddi & Maria Grazia Orofino
You can also search for this author in PubMed Google Scholar
Contributions
OM, GC conceptualized and designed the study; OM, GC, EP, CT, VF, SB, AP, MGO, AV, GLN treated patients, collected and assembled the data; OM and FE performed the statistical analysis; OM, GC wrote the paper; OM, GC, FE, EP, CT, VF, SB, AP, MGO, AV, GLN were responsible for the final approval of the paper.
Corresponding author
Correspondence to Giovanni Caocci .
Ethics declarations
Competing interests.
FE: consultancy fees from Amgen, Abbvie, Janssen, and Novartis. Research support (Institution) from Abbvie, Amgen, Novartis, all unrelated to this work. The other authors have no competing interests to disclose.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplemental material, rights and permissions.
Reprints and permissions
About this article
Cite this article.
Mulas, O., Caocci, G., Efficace, F. et al. Long-term health-related quality of life in patients with β-thalassemia after unrelated hematopoietic stem cell transplantation. Bone Marrow Transplant 57 , 1833–1836 (2022). https://doi.org/10.1038/s41409-022-01823-2
Download citation
Received : 15 June 2022
Revised : 31 August 2022
Accepted : 01 September 2022
Published : 16 September 2022
Issue Date : December 2022
DOI : https://doi.org/10.1038/s41409-022-01823-2
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies

Click through the PLOS taxonomy to find articles in your field.
For more information about PLOS Subject Areas, click here .
Thalassemia
- Autosomal recessive diseases
- Hemoglobinopathies
- Alpha-thalassemia
- Beta-thalassemia
- Get an email alert for Thalassemia
- Get the RSS feed for Thalassemia
Showing 1 - 13 of 63
View by: Cover Page List Articles
Sort by: Recent Popular
Two risk factors for hypozincemia in diabetic β-thalassemia patients: Hepatitis C and deferasirox
Hadi Darvishi-Khezri, Hossein Karami, [ ... ], Hasan Karami
Economic burden of Thalassemia treatment: An analysis from the Vietnam Social Security perspective
Hoang-Thy Nhac-Vu, Van Thi-Ngoc Tran, [ ... ], Tho Le
0 -thalassemia-- SEA ,-- THAI , and-- CR type deletions">Single-tube multiplex real-time PCR with EvaGreen and high-resolution melting analysis for diagnosis of α 0 -thalassemia-- SEA ,-- THAI , and-- CR type deletions
Chedtapak Ruengdit, Manoo Punyamung, Nutjeera Intasai, Sakorn Pornprasert
Speckle tracking echocardiography-derived left ventricular global longitudinal strain in ex-thalassaemics
Amal Paul, Uday Kulkarni, [ ... ], Alok Srivastava
Knowledge and attitudes of thalassaemia among high-risk indigenous university students in Bangladesh: A pilot study
Md. Mahbub Hasan, Khaza Md. Kapil Uddin, [ ... ], Mohammad Sorowar Hossain
Anemia in an ethnic minority group in lower northern Thailand: A community-based study investigating the prevalence in relation to inherited hemoglobin disorders and iron deficiency
Thinzar Win Pyae, Kanokwan Sanchaisuriya, [ ... ], Supan Fucharoen
Frequency of unnecessary prenatal diagnosis of hemoglobinopathies: A large retrospective analysis and implication to improvement of the control program
Kritsada Singha, Supawadee Yamsri, [ ... ], Supan Fucharoen
Integration of targeted sequencing and pseudo-tetraploid genotyping into clinically assisted decision support for β-thalassemia invasive prenatal diagnosis
Wenguang Jia, Jiying Shi, [ ... ], Ping Chen

Electrophysiological properties and heart rate variability of patients with thalassemia major in Jakarta, Indonesia
Rubiana Sukardi, Pustika Amalia Wahidiyat, [ ... ], Mulyadi M. Djer
0 -thalassemia: Application to population screening and prenatal diagnosis">Loop-mediated isothermal amplification (LAMP) colorimetric phenol red assay for rapid identification of α 0 -thalassemia: Application to population screening and prenatal diagnosis
Wittaya Jomoui, Hataichanok Srivorakun, Siriyakorn Chansai, Supan Fucharoen
Pediatric thalassemic patients have higher incidence of asthma: A nationwide population-based retrospective cohort study
Hsin-Yi Hsieh, Lin-Chi Huang, [ ... ], Jiunn-Ming Sheen
Hepcidin induces intestinal calcium uptake while suppressing iron uptake in Caco-2 cells
Supathra Phoaubon, Kornkamon Lertsuwan, Jarinthorn Teerapornpuntakit, Narattaphol Charoenphandhu
Epidemiology of thalassemia among the hill tribe population in Thailand
Tawatchai Apidechkul, Fartima Yeemard, [ ... ], Ratipark Tamornpark
Connect with Us
- PLOS ONE on Twitter
- PLOS on Facebook
Articles About Thalassemia
Key findings, featured articles, scientific articles.
Study Finds Methods for Alpha-Thalassemia Screening and Reporting Vary Across Newborn Screening Programs This study is about current screening practices to detect alpha thalassemia.
Study Highlights Opportunity to Improve Knowledge of and Familiarity with Thalassemia among Variety of California Healthcare Providers The increase in number of patients with thalassemia living in California highlights the importance of provider knowledge about thalassemia in order to effectively serve these patients in their communities.
Thalassemia Awareness By staying committed to long-term treatment, people with thalassemia can enjoy a full life.
- Patterson S, Singleton A, Branscomb J, Nsonwu V, Spratling R. Transfusion complications in thalassemia: patient knowledge and perspectives . Frontiers in Medicine, Hematology. 2022; 9:772886.
- Lal A, Wong T, Keel S, et al. The transfusion management of beta thalassemia in the United States . Transfusion. 2021;61(10):3027-3039. doi:10.1111/trf.16640
- Mensah C, Sheth S. Optimal strategies for carrier screening and prenatal diagnosis of α- and β-thalassemia. Hematology Am Soc Hematol Educ Program. 2021;2021(1):607-613. doi:10.1182/hematology.2021000296
- Rollins MR, Boudreaux J, Eckman J, Branscomb J, Zhou M, Snyder A. Intermittent transfusions for treatment of thalassemia in the state of Georgia, 2007-2016 . Annals of Hematology and Oncology. 2021; 8(4): 1340.
- Bender M, Yusuf C, Davis T, et al. Newborn Screening Practices and Alpha-Thalassemia Detection — United States, 2016 . MMWR Morb Mortal Wkly Rep 2020;69:1269–1272.
- Radke T, Paulukonis S, Hulihan M, Feuchtbaum L. Providers’ Perspectives on Treating Patients with Thalassemia. JPediatr Hematol Oncol. 2019 Jul 25. Doi:10.1097/MPH. 0000000000001573. [Epub ahead of print]
- Fasano RM, Branscomb J, Lane PA, Josephson CD, Snyder AB, Eckman JR. Transfusion service knowledge and immunohaematological practices related to sickle cell disease and thalassemia Transfus Med. 2019 Feb 10.
- Spratling R, Lawrence RH. Facilitators and Barriers to Minority Blood Donations: A Systematic Review Nurs Res. 2019 Mar 1. [Epub ahead of print]
- Lal A, Wong TE, Andrews J, Balasa VV, Chung JH, Forester CM, et al. Transfusion practices and complications in thalassemia . Transfusion 2018 Sep 27. [Epub ahead of print]
- Singleton A, Spratling R. A strategic planning tool for increasing African American blood donation . Health Promot Pract . 2018 May. [Epub ahead of print]
- Mainous AG 3rd, Carek PJ, Lynch K, Tanner RJ, Hulihan MM, Baskin J, et al. Effectiveness of clinical decision support based intervention in the improvement of care for adult sickle cell disease patients in primary care . J Am Board Fam Med . 2018;31(5):812-816.
- Fasano RM, Meyer EK, Branscomb J, White MS, Gibson RW, Eckman JR. Impact of red blood cell antigen matching on alloimmunization and transfusion complications in patients with sickle cell disease: A systematic review. Transfus Med Rev . 2018 Jul 26. [Epub ahead of print]
- Yee MEM, Josephson CD, Winkler AM, Webb J, Luban NLC, Leong T, et al. Hemoglobin A clearance in children with sickle cell anemia on chronic transfusion therapy . Transfusion 2018;58(6):1363-1371.
- Yee MEM, Josephson CD, Winkler AM, Webb J, Luban NLC, Leong T, et al. Red blood cell minor antigen mismatches during chronic transfusion therapy for sickle cell anemia . Transfusion 2017;57(11):2738-2746.
To view additional recent articles on thalassemia, please see Cooley’s Anemia: 9th Symposium , sponsored by the New York Academy of Sciences and Cooley’s Anemia Foundation.
- Deep Vein Thrombosis (DVT) and Clotting
- Sickle Cell Disease
- Women’s Blood Disorders
- CDC’s National Center on Birth Defects and Developmental Disabilities

Join the Public Health Webinar Series on Blood Disorders
- Upcoming webinar
- View past webinars
Exit Notification / Disclaimer Policy
- The Centers for Disease Control and Prevention (CDC) cannot attest to the accuracy of a non-federal website.
- Linking to a non-federal website does not constitute an endorsement by CDC or any of its employees of the sponsors or the information and products presented on the website.
- You will be subject to the destination website's privacy policy when you follow the link.
- CDC is not responsible for Section 508 compliance (accessibility) on other federal or private website.
- Introduction to Genomics
- Educational Resources
- Policy Issues in Genomics
- The Human Genome Project
- Funding Opportunities
- Funded Programs & Projects
- Division and Program Directors
- Scientific Program Analysts
- Contact by Research Area
- News & Events
- Research Areas
- Research investigators
- Research Projects
- Clinical Research
- Data Tools & Resources
- Genomics & Medicine
- Family Health History
- For Patients & Families
- For Health Professionals
- Jobs at NHGRI
- Training at NHGRI
- Funding for Research Training
- Professional Development Programs
- NHGRI Culture
- Social Media
- Broadcast Media
- Image Gallery
- Press Resources
- Organization
- NHGRI Director
- Mission & Vision
- Policies & Guidance
- Institute Advisors
- Strategic Vision
- Leadership Initiatives
- Diversity, Equity, and Inclusion
- Partner with NHGRI
- Staff Search
About Thalassemia
Thalassemia is a group of inherited diseases of the blood that affect a person's ability to produce hemoglobin, resulting in anemia.
What do we know about heredity and thalassemia?
Thalassemia is actually a group of inherited diseases of the blood that affect a person's ability to produce hemoglobin, resulting in anemia. Hemoglobin is a protein in red blood cells that carries oxygen and nutrients to cells in the body. About 100,000 babies worldwide are born with severe forms of thalassemia each year. Thalassemia occurs most frequently in people of Italian, Greek, Middle Eastern, Southern Asian and African Ancestry.
The two main types of thalassemia are called "alpha" and "beta," depending on which part of an oxygen-carrying protein in the red blood cells is lacking. Both types of thalassemia are inherited in the same manner. The disease is passed to children by parents who carry the mutated thalassemia gene. A child who inherits one mutated gene is a carrier, which is sometimes called "thalassemia trait." Most carriers lead completely normal, healthy lives.
A child who inherits two thalassemia trait genes - one from each parent - will have the disease. A child of two carriers has a 25 percent chance of receiving two trait genes and developing the disease, and a 50 percent chance of being a thalassemia trait carrier.
Most individuals with alpha thalassemia have milder forms of the disease, with varying degrees of anemia. The most severe form of alpha thalassemia, which affects mainly individuals of Southeast Asian, Chinese and Filipino ancestry, results in fetal or newborn death.
A child who inherits two copies of the mutated gene for beta thalassemia will have beta thalassemia disease. The child can have a mild form of the disease, known as thalassemia intermedia, which causes milder anemia that rarely requires transfusions.
Thalassemia Major: A Serious Disorder
The more severe form of the disease is thalassemia major, also called Cooley's Anemia. It is a serious disease that requires regular blood transfusions and extensive medical care.
Those with thalassemia major usually show symptoms within the first two years of life. They become pale and listless and have poor appetites. They grow slowly and often develop jaundice. Without treatment, the spleen, liver and heart soon become greatly enlarged. Bones become thin and brittle. Heart failure and infection are the leading causes of death among children with untreated thalassemia major.
The use of frequent blood transfusions and antibiotics has improved the outlook for children with thalassemia major. Frequent transfusions keep their hemoglobin levels near normal and prevent many of the complications of the disease. But repeated blood transfusions lead to iron overload - a buildup of iron in the body - that can damage the heart, liver and other organs. Drugs known as "iron chelators" can help rid the body of excess iron, preventing or delaying problems related to iron overload.
Thalassemia has been cured using bone marrow transplants. However, this treatment is possible only for a small minority of patients who have a suitable bone marrow donor. The transplant procedure itself is still risky and can result in death.
Gene Therapy Offers Hope for a Cure
Scientists are working to develop a gene therapy that may offer a cure for thalassemia. Such a treatment might involve inserting a normal beta globin gene (the gene that is abnormal in this disease) into the patient's stem cells, the immature bone marrow cells that are the precursors of all other cells in the blood.
Another form of gene therapy could involve using drugs or other methods to reactivate the patient's genes that produce fetal hemoglobin - the form of hemoglobin found in fetuses and newborns. Scientists hope that spurring production of fetal hemoglobin will compensate for the patient's deficiency of adult hemoglobin.
Is there a test for thalassemia?
Blood tests and family genetic studies can show whether an individual has thalassemia or is a carrier. If both parents are carriers, they may want to consult with a genetic counselor for help in deciding whether to conceive or whether to have a fetus tested for thalassemia.
Prenatal testing can be done around the 11th week of pregnancy using chorionic villi sampling (CVS). This involves removing a tiny piece of the placenta. Or, the fetus can be tested with amniocentesis around the 16th week of pregnancy. In this procedure, a needle is used to take a sample of the fluid surrounding the baby for testing.
Assisted reproductive therapy is also an option for carriers who don't want to risk giving birth to a child with thalassemia. A new technique, pre-implantation genetic diagnosis (PGD), used in conjunction with in vitro fertilization, may enable parents who have thalassemia or carry the trait to give birth to healthy babies. Embryos created in-vitro are tested for the thalassemia gene before being implanted into the mother, allowing only healthy embryos to be selected.
Additional Resources for Thalassemia
NHLBI: Thalassemias
Medline Plus: Thalassemia
Genetics Home Reference: Beta thalassemia
Thalassemia.com
GARD: Thalassemia
Last updated: December 27, 2013
IMAGES
VIDEO
COMMENTS
Thalassemia is an inherited disease, meaning that at least one of the parents must be a carrier for the disease. It is caused by either a genetic mutation or a deletion of certain key gene fragments. Alpha thalassemia is caused by alpha-globin gene deletion which results in reduced or absent production of alpha-globin chains. Alpha globin gene ...
Thalassaemia is a diverse group of genetic disorders with a worldwide distribution affecting globin chain synthesis. The pathogenesis of thalassaemia lies in the unbalanced globin chain production, leading to ineffective erythropoiesis, increased haemolysis, and deranged iron homoeostasis. The clinical phenotype shows heterogeneity, ranging from close to normal without complications to severe ...
Molecular Pathogenesis. β-Thalassemia is caused by mutations resulting in a single nucleotide substitution, small deletions or insertions within the β-globin gene or its immediate flanking ...
To evaluate the prevalence and the performance of routine surveillance for thalassemia related complications during 2 periods; before and after published CPGs (2012-2014 vs 2015-2017), data ...
β-Thalassemia results from insufficient production of the hemoglobin subunit β-globin (β +) or from the absence of β-globin (β 0 ). Low levels of adult hemoglobin (HbA, or α 2 β 2) are ...
Editorial on the Research Topic Insights in thalassemia: from genomics to clinical practice. Thalassemia, a common inherited autosomal recessive disorder, is characterized by inefficient or absent hemoglobin synthesis, resulting in various severities of anemia ().Though considered a global medical and public health concern, its greatest impact is clearly felt in countries with limited ...
β-thalassemia is a disease caused by genetic mutations including a nucleotide change, small insertions or deletions in the β-globin gene, or in rare cases, gross deletions into the β-globin gene. These mutations affect globin-chain subunits within the hemoglobin tetramer what induces an imbalance in the α/β-globin chain ratio, with an excess of free α-globin chains that triggers the most ...
DOI: 10.1056/NEJMra1404415. The thalassemias are the most common human monogenic diseases. 1 These inherited disorders of hemoglobin synthesis are characterized by a reduced production of globin ...
Review Article. ISSN No. 2394-3971. Abstract. Thalassemia's ar e genetic disorders inherited from a person's parents. Thalassemia's are. prevalent worldwide with 25,000 deaths in 2013 ...
Credit: toeytoey2530/ iStock / Getty Images Plus. Adding a functional gene to defective blood stem cells is a successful therapy for patients with severe beta thalassemia, according to an ...
This narrative review was performed by collecting clinical trials, primary research, and reviews on molecular genetics and prospects for β-thalassemia therapy. Articles published in peer-reviewed scientific journals were also included. Articles were excluded if they were not written in English or published in peer-reviewed scientific journals.
In the time period considered in the research (1995-2016) 127 patients were transplanted (60 matched unrelated and 67 sibling HSCT) with 81% of Overall Survival (OS) and 77% of Thalassemia Free ...
Frequency of unnecessary prenatal diagnosis of hemoglobinopathies: A large retrospective analysis and implication to improvement of the control program. Kritsada Singha, Supawadee Yamsri, Integration of targeted sequencing and pseudo-tetraploid genotyping into clinically assisted decision support for β-thalassemia invasive prenatal diagnosis ...
The increase in number of patients with thalassemia living in California highlights the importance of provider knowledge about thalassemia in order to effectively serve these patients in their communities. Featured Articles. Thalassemia Awareness By staying committed to long-term treatment, people with thalassemia can enjoy a full life.
728 n engl j med 384;8 nejm.org February 25, 2021 The new england journal of medicine β-thalassemia are closely tied to the degree of im-balance between α-globin and β-globin chains. Deficient ...
The National Institutes of Health-sponsored North American Thalassemia Clinical Research Network (TCRN) conducted a cross-sectional study of 721 patients with thalassemia syndromes. A detailed ...
Thalassemia is actually a group of inherited diseases of the blood that affect a person's ability to produce hemoglobin, resulting in anemia. Hemoglobin is a protein in red blood cells that carries oxygen and nutrients to cells in the body. About 100,000 babies worldwide are born with severe forms of thalassemia each year.