- Sickle cell anemia

On this page
When to see a doctor, risk factors, complications.
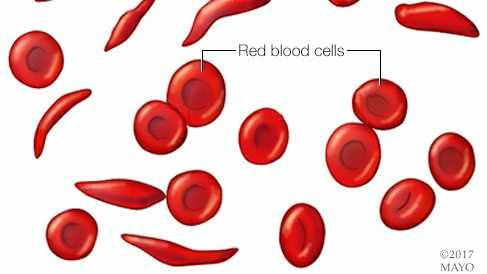
Sickle cell anemia is one of a group of inherited disorders known as sickle cell disease. It affects the shape of red blood cells, which carry oxygen to all parts of the body.
Red blood cells are usually round and flexible, so they move easily through blood vessels. In sickle cell anemia, some red blood cells are shaped like sickles or crescent moons. These sickle cells also become rigid and sticky, which can slow or block blood flow.
The current approach to treatment is to relieve pain and help prevent complications of the disease. However, newer treatments may cure people of the disease.

Red blood cells are usually round and flexible. In sickle cell anemia, some red blood cells look like sickles used to cut wheat. These unusually shaped cells give the disease its name.
Products & Services
- A Book: Mayo Clinic Family Health Book, 5th Edition
- Newsletter: Mayo Clinic Health Letter — Digital Edition
Symptoms of sickle cell anemia usually appear around 6 months of age. They vary from person to person and may change over time. Symptoms can include:
- Anemia. Sickle cells break apart easily and die. Typical red blood cells usually live for about 120 days before they need to be replaced. But sickle cells usually die in 10 to 20 days, leaving a shortage of red blood cells. This is known as anemia. Without enough red blood cells, the body can't get enough oxygen. This causes fatigue.
Episodes of pain. Periodic episodes of extreme pain, called pain crises, are a major symptom of sickle cell anemia. Pain develops when sickle-shaped red blood cells block blood flow through tiny blood vessels to the chest, abdomen and joints.
The pain varies in intensity and can last for a few hours to a few days. Some people have only a few pain crises a year. Others have a dozen or more a year. A severe pain crisis requires a hospital stay.
Some people with sickle cell anemia also have chronic pain from bone and joint damage, ulcers, and other causes.
- Swelling of hands and feet. Sickle-shaped red blood cells block blood circulation in the hands and feet, which can cause them to swell.
- Frequent infections. The spleen is important for protecting against infections. Sickle cells can damage the spleen, raising the risk of developing infections. Babies and children with sickle cell anemia commonly receive vaccinations and antibiotics to prevent potentially life-threatening infections, such as pneumonia.
- Delayed growth or puberty. Red blood cells provide the body with the oxygen and nutrients needed for growth. A shortage of healthy red blood cells can slow growth in babies and children and delay puberty in teenagers.
- Vision problems. Tiny blood vessels that supply blood to the eyes can become plugged with sickle cells. This can damage the portion of the eye that processes visual images, called the retina, and lead to vision problems.
See your healthcare professional right away if you or your child has symptoms of sickle cell anemia, including fever or stroke.
Infections often start with a fever and can be life-threatening. Because children with sickle cell anemia are prone to infections, seek prompt medical attention for a fever greater than 101.5 degrees Fahrenheit (38.5 degrees Celsius).
Seek emergency care for symptoms of stroke, which include:
- One-sided paralysis or weakness in the face, arms or legs.
- Difficulty walking or talking.
- Sudden vision changes.
- Unexplained numbness.
- Severe headache.
From Mayo Clinic to your inbox
Sickle cell anemia is caused by a change in the gene that tells the body to make hemoglobin. Hemoglobin is the iron-rich compound in red blood cells that allows these cells to carry oxygen from the lungs to the rest of the body. The hemoglobin associated with sickle cell anemia causes red blood cells to become rigid, sticky and misshapen.
For a child to have sickle cell anemia, both parents must carry one copy of the sickle cell gene and pass both copies to the child.
If only one parent passes the sickle cell gene to the child, that child will have the sickle cell trait. With one typical hemoglobin gene and one sickle cell gene, people with the sickle cell trait make both typical hemoglobin and sickle cell hemoglobin.
Their blood might contain some sickle cells, but they generally don't have symptoms. They're carriers of the disease. That means they can pass the gene to their children.
For a baby to have sickle cell anemia, both parents must carry a sickle cell gene. In the United States, sickle cell anemia most commonly affects people of African, Mediterranean and Middle Eastern descent.
Sickle cell anemia can lead to a host of complications, including:
- Stroke. Sickle cells can block blood flow to the brain. Signs of stroke include seizures, weakness or numbness of the arms and legs, sudden speech difficulties, and loss of consciousness. If your child has any of these signs or symptoms, seek medical treatment right away. A stroke can be fatal.
- Acute chest syndrome. A lung infection or sickle cells blocking blood vessels in the lungs can cause this life-threatening complication. Symptoms include chest pain, fever and difficulty breathing. Acute chest syndrome might need emergency medical treatment.
- Avascular necrosis. Sickle cells can block the blood vessels that supply blood to the bones. When the bones don't get enough blood, joints may narrow and bones can die. This can happen anywhere but most often happens in the hip.
- Pulmonary hypertension. People with sickle cell anemia can develop high blood pressure in their lungs. This complication usually affects adults. Shortness of breath and fatigue are common symptoms of this condition, which can be fatal.
- Organ damage. Sickle cells that block blood flow to organs deprive the affected organs of blood and oxygen. In sickle cell anemia, blood also is low in oxygen. This lack of oxygen-rich blood can damage nerves and organs, including the kidneys, liver and spleen, and can be fatal.
- Splenic sequestration. Sickle cells can get trapped in the spleen, causing it to enlarge. This may cause abdominal pain on the left side of the body and can be life-threatening. Parents of children with sickle cell anemia can learn how to locate and feel their child's spleen for enlargement.
- Blindness. Sickle cells can block tiny blood vessels that supply blood to the eyes. Over time, this can lead to blindness.
- Leg ulcers. Sickle cell anemia can cause painful open sores on the legs.
- Gallstones. The breakdown of red blood cells produces a substance called bilirubin. A high level of bilirubin in the body can lead to gallstones.
- Priapism. Sickle cell anemia can cause painful, long-lasting erections, known as priapism. Sickle cells can block the blood vessels in the penis, which can lead to impotence over time.
- Deep vein thrombosis. Sickled red blood cells can cause blood clots, increasing the risk of a clot lodging in a deep vein, known as deep vein thrombosis. It also increases the risk of a blood clot lodging in a lung, known as pulmonary embolism. Either can cause serious illness or even death.
- Pregnancy complications. Sickle cell anemia can increase the risk of high blood pressure and blood clots during pregnancy. It also can increase the risk of miscarriage, premature birth and low birth weight babies.
If you carry the sickle cell trait, it can help to see a genetic counselor before you get pregnant. A counselor can help you understand your risk of having a child with sickle cell anemia. You also can learn about possible treatments, preventive measures and reproductive options.
Dec 22, 2023
- Sickle cell disease. National Heart, Lung, and Blood Institute. https://www.nhlbi.nih.gov/health-topics/sickle-cell-disease. Accessed Aug. 4, 2023.
- Field JJ, et al. Overview of the management and prognosis of sickle cell disease. https://www.uptodate.com/contents/search. Accessed Aug. 4, 2023.
- AskMayoExpert. Sickle cell disease. Mayo Clinic; 2022.
- Sickle cell disease (SCD). Centers for Disease Control and Prevention. https://www.cdc.gov/ncbddd/sicklecell/index.html. Accessed Aug. 4, 2023.
- Hoffman R, et al. Pain Management and Antiemetic Therapy in Hematologic Disorders. In: Hematology: Basic Principles and Practice. 8th ed. Elsevier; 2023. https://www.clinicalkey.com. Accessed Aug. 4, 2023.
- Ferri FF. Sickle cell disease. In: Ferri's Clinical Advisor 2024. Elsevier; 2024. https://www.clinicalkey.com. Accessed Aug. 4, 2023.
- Lyfgenia (prescribing information). Bluebird Bio; 2023. https://www.fda.gov/vaccines-blood-biologics/lyfgenia. Accessed Dec. 11, 2023.
- Casgevy (prescribing information). Vertex Pharmaceuticals; 2023. https://www.fda.gov/vaccines-blood-biologics/casgevy. Accessed Dec. 11, 2023.
- Diseases & Conditions
- Sickle cell anemia symptoms & causes
News from Mayo Clinic
More Information
Associated procedures.
- Blood transfusion
- Bone marrow transplant
CON-XXXXXXXX
Your gift holds great power – donate today!
Make your tax-deductible gift and be a part of the cutting-edge research and care that's changing medicine.

BARBARA P. YAWN, MD, MSc, MSPH, AND JOYLENE JOHN-SOWAH, MD, MPH
Am Fam Physician. 2015;92(12):1069-1076A
Patient information : A handout on this topic is available at https://familydoctor.org/familydoctor/en/diseases-conditions/sickle-cell-disease.html .
Author disclosure: No relevant financial affiliations.
Family physicians are the primary and sometimes only health care resource for families affected by sickle cell disease. Recently published guidelines provide important recommendations for health maintenance, acute care, and monitoring of disease-modifying therapy in persons with this condition. This overview highlights some of the most important clinical activities that can and should be carried out in the community care setting. Children with sickle cell anemia should receive prophylactic penicillin from birth through at least five years of age, and all persons with sickle cell disease require vaccination to prevent invasive pneumococcal disease. Annual screening with transcranial Doppler ultrasonography is recommended for all children with sickle cell disease beginning at two years of age and continuing through adolescence to evaluate the risk of stroke and to initiate transfusion therapy in those at high risk. Vasoocclusive crises require immediate and adequate analgesia appropriate to the level of patient-reported pain. Antibiotics, hospitalization, and incentive spirometry are indicated for those with acute chest syndrome. There is strong evidence to support the promotion and use of hydroxyurea therapy in patients nine months and older who have sickle cell anemia because its use can decrease the frequency of vasoocclusive crises and acute chest syndrome with limited adverse effects.
Family physicians and family medical homes are essential to the care of children, adults, and families affected by sickle cell disease (SCD). Many of the approximately 100,000 U.S. residents who have SCD do not have regular or convenient access to comprehensive SCD centers. This summary of the recently published National Institutes of Health–sponsored SCD guidelines, Evidence-Based Management of Sickle Cell Disease , Expert Panel Report 2014, 1 is intended to support, enhance, and expand the knowledge of basic aspects of care for patients with SCD. Specifically highlighted are several clinical actions to enhance preventive care, manage some of the most common acute and chronic complications of SCD, and initiate and monitor the two SCD-specific disease-modifying therapies, hydroxyurea and chronic blood transfusion therapy.
WHAT IS NEW ON THIS TOPIC: SICKLE CELL DISEASE
Offer hydroxyurea therapy to infants, children, and adolescents with sickle cell anemia regardless of clinical severity to reduce sickle cell disease–related complications.
Use an individualized prescribing and monitoring protocol or a sickle cell disease–specific protocol whenever possible to promote rapid, effective, and safe analgesic management and resolution of vasoocclusive crises in children and adults.
Overview of SCD
SCD is a genetic disorder that results in the formation of sickled red blood cells (RBCs). Patients with SCD include those who are homozygous for sickle hemoglobin (HbSS, also called sickle cell anemia [SCA]) and those with one sickle hemoglobin gene plus a gene for another abnormal hemoglobin type (e.g., HbSβ + -thalassemia, HbSC). The recommendations in these guidelines do not apply to persons who have sickle cell trait (also called carriers [one gene for HbS and another for normal HbA]). SCD predominantly affects persons of African ancestry; a minority of affected persons are of Hispanic, Middle Eastern, or Asian Indian descent. Deoxygenated RBCs containing predominantly HbS develop a sickle or crescent shape, become inflexible, increase blood viscosity, and block or limit blood flow within limbs or organs. 2 The process is further aggravated by abnormal interactions of these RBCs with leukocytes, platelets, vascular endothelium, and clotting factors, 3 , 4 thus causing acute and chronic complications. The frequency and severity of complications can be reduced with hydroxyurea or blood transfusion therapy. 5 – 8 However, both treatments are currently underutilized 5 – 8 and are not commonly initiated or monitored by family physicians.
Caring for Patients with SCD in the Community Care Setting
Screening, prevention, and immunizations.
Many patients with SCD do not receive routine preventive care recommended by the U.S. Preventive Services Task Force and immunizations recommended by the Advisory Committee on Immunization Practices. Persons with SCD also benefit from specialized condition-specific preventive strategies. Children with SCA are at increased risk of invasive pneumococcal disease. Therefore, in addition to the recommended pneumococcal vaccinations for all infants and children, those with SCA should receive prophylactic oral penicillin (125 mg twice daily for children younger than three years; 250 mg twice daily for those three years and older) once the diagnosis is established; this regimen should be continued until at least five years of age. 9 – 11
Persons with SCD are also at increased risk of vascular complications, particularly stroke. All persons with SCA should be screened annually with transcranial Doppler ultrasonography (TCD) beginning at two years of age and continuing until at least 16 years of age. 12 – 14 TCD measures and reports the average velocity of blood flow through the internal carotid and proximal middle cerebral arteries. A TCD finding of 200 cm per second or greater correlates with a 10% annual increase in stroke risk. 7 When TCD findings are marginal or conditional (170 to 199 cm per second) or elevated (200 cm per second or greater), the child is considered a candidate for blood transfusion therapy to prevent stroke and should be referred for comanagement with a subspecialist. 15 , 16
Recommendations for these and other common conditions are summarized in Table 1 . 1
In women with SCD, regular use of contraception can decrease the health risks associated with an unintended pregnancy. Progestin-only contraceptives (pills, injections, and implants) and barrier methods have no restrictions or concerns for use in women with SCD. 17 , 18 The levonorgestrel-releasing intrauterine system is also associated with few risks, except for those associated with the device placement. 17 , 18 Pregnancy in women with SCD is associated with preterm delivery, preeclampsia, stillbirth, high maternal mortality, and severe fetal anemia. Extra prenatal surveillance is recommended in consultation with a maternal-fetal subspecialist whenever possible. 19 , 20 Both men and women with SCD require basic contraceptive information and referral for genetic counseling when making reproductive decisions. 21
COMMON ACUTE AND CHRONIC COMPLICATIONS
Persons with SCD can have acute complications that require rapid interventions to avert or lessen the risk of life-threatening consequences. The most common complication is a vasoocclusive crisis (VOC). These episodes usually present with sudden onset of severe pain, often localized to the extremities, chest, or back, but with few objective findings. Nearly all persons with SCD will have a VOC during their lifetime, 22 , 23 sometimes as early as six months of age, when it presents as dactylitis. Recurrences have variable presentations and frequency. 23 , 24 VOC is a clinical diagnosis with no objective diagnostic tests. Other potential causes for the acute pain should be excluded while providing required analgesia. Primary management of VOC includes rapid triage, assessment, and administration of appropriate analgesics. Pain management should be based on patient-reported pain severity. For mild or moderate pain, treatment may begin with nonsteroidal anti-inflammatory drugs, if they are not contraindicated. For more severe pain, opioids are the first-line therapy; they should be provided immediately and therapy should be guided by the patient's history of previous successful regimens. 25 Meperidine (Demerol) should not be used unless it is the only effective alternative available. 26 Hydration and other nonpharmacologic therapies (e.g., maintaining body temperature) may also be useful.
Concerns about drug-seeking behavior are pervasive and often triggered by patients with SCD who have frequent VOCs that require potent analgesia. These concerns should be addressed after adequately treating the acute pain. The use of an individualized VOC therapy plan that is carried with the patient and presented at urgent, emergency, or other care sites may lessen these concerns and facilitate rapid VOC management ( Figure 1 ) . 1 Evaluation of suspected or possible drug-seeking behavior requires careful review of previous drug use, discussions with the patient's prescribing physicians, and objective information about drug abuse behavior. Consultation with a behavioral health expert is indicated when there are concerns about pain medication abuse. 27
Acute chest syndrome (ACS), defined as the presence of a new lung infiltrate in a patient with acute onset of lower respiratory symptoms such as cough and shortness of breath, is less common than VOC but potentially life threatening. In children, it often presents with fever and signs of middle lobe lung involvement, whereas adults are often afebrile and have multilobe infiltrates. ACS can present on its own or as a complication of a VOC; it requires prompt evaluation and, once diagnosed, early intervention with antibiotic therapy and hospitalization. 28 , 29 During hospitalization for a VOC, incentive spirometry can reduce the risk of ACS. 30 When evaluating a new lung infiltrate in a patient with SCD, physicians should consider ACS before assuming the infiltrate represents community-acquired pneumonia.
Fever greater than 101°F (38.3°C), even in the absence of other signs of infection, should be evaluated carefully. Because of absent or reduced splenic function, persons with SCD have a high risk of overwhelming bacterial infections or sepsis. 31 Fevers with possible infections should be treated empirically until culture results are available ( Table 2 ) . 1
Hepatobiliary tract complications (e.g., cholelithiasis, acute cholecystitis, biliary sludge, acute choledocholithiasis) are common in persons with SCD, particularly in those with SCA. Recommended clinical responses are similar to those in patients without SCD who have these conditions. Gallstones occur in up to 75% of adults with SCD and, when asymptomatic, should be managed with watchful waiting. 27
Disease-Modifying Therapies
Hydroxyurea therapy decreases SCD-related complications but is currently underused. Family physicians can be key to increasing the use of this therapy; they should ensure that every person affected by SCD is informed of the potential benefits and risks of hydroxyurea therapy, and should learn to initiate and monitor it.
Hydroxyurea works primarily by increasing the level of fetal hemoglobin (HbF), which does not sickle. This improves several clinical outcomes, such as decreasing the frequency of VOCs and ACS, reducing mortality, and decreasing the need for RBC transfusions and hospitalizations. 32 – 34 Hydroxyurea is rapidly absorbed, has nearly complete bioavailability, and requires only once-daily oral dosing.
All persons nine months and older who have SCA are candidates for hydroxyurea therapy, although those with compound heterozygous conditions (e.g., HbSC, HbSβ + -thalassemia) require therapy less often than those with HbSS. Although the seminal randomized controlled trial of hydroxyurea therapy enrolled only adults with a history of moderate to severe VOCs, results from observational studies prompted the Expert Panel to broaden the recommendations to include adults with frequent VOCs or symptomatic anemia ( Table 3 ) . 1 More recent studies included infants and young children without a history of VOCs, resulting in the recommendation to offer the therapy to even asymptomatic infants with SCA beginning at nine months of age. 35 – 37 eTable A presents a consensus plan for initiating and monitoring hydroxyurea therapy.
Longer-term observational studies suggest that hydroxyurea therapy has long-term beneficial effects across all age groups with limited adverse effects—usually myelotoxicity in the form of reversible cytopenias, but no deleterious effects on growth and development. Thus far, there is no evidence of increased carcinogenicity or mutagenesis during long-term therapy in adults. 38 However, hydroxyurea therapy is contraindicated in pregnant and breastfeeding women. 27
Donor RBC transfusion, the first disease-modifying therapy used for SCD, reduces the percentage of circulating RBCs with HbS, thereby treating acute symptomatic anemia as well as treating and preventing many SCD complications. However, the need for repeated venous access and the associated risks and complications, such as alloimmunization and iron overload, limit its use. 39 , 40
Donor RBCs may be administered as a simple transfusion—without removal of any recipient blood—or as an exchange transfusion, which involves removal of recipient blood before and possibly during infusion. Transfusions can be episodic or long term. Episodic transfusion is used acutely in response to an SCD complication or prophylactically in preparation for general anesthesia and surgery. Long-term transfusion therapy is used when sustained reduction of HbS (e.g., to less than 30%) is desired for primary or secondary prophylaxis for specific SCD complications, most commonly stroke in children. 14 – 16
Table 4 summarizes recommendations for and against the use of transfusion therapy for SCD and its complications. 1
Chronic blood transfusion programs are usually based in comprehensive SCD centers, but benefit from linkages to physicians who provide community continuity of care. Within the center, careful blood matching, including RBC phenotype matching to at least the C, E, and K antigens, decreases the incidence of alloimmunization and transfusion reactions. Detailed monitoring strategies must be used and continued into community practices to identify delayed transfusion reactions, which can present as acute anemia, pain, or jaundice days to weeks later; to maintain low HbS levels; and to track and treat transfusion-related iron overload. 40 The Expert Panel supports quarterly monitoring of serum ferritin levels. Chelation therapy can effectively remove excess iron in patients with confirmed iron overload. 27
Resources for Consultation
Family physicians may have limited experience in offering continuing or emergency care to patients with SCD, which makes it important to learn about consultative resources available regionally or nationally. Comanagement with adult or pediatric hematology-oncology subspecialists can enhance the family physician's knowledge and overall care of patients with SCD. In special cases (e.g., stroke, priapism, ACS), assistance can be garnered from other local subspecialists. However, local or regional subspecialists may also have limited experience in the care of SCD-related complications. Care of patients with SCD usually requires the involvement of a large group of health care professionals.
Data Sources : This is a summary of and highlights from evidence-based guidelines. We did not do any further searches than those completed for the guidelines. The methods for guideline development are described within the full guideline text ( http://www.nhlbi.nih.gov/health-pro/guidelines/sickle-cell-disease-guidelines/index.htm ). In summary, key questions (n > 40) were developed to guide selection of keywords used to search the available English-language literature from the Cochrane Database of Systematic Reviews, Bandolier, and the National Guideline Clearinghouse, and recommendations posted by the U.S. Preventive Services Task Force and the Advisory Committee on Immunization Practices.
The Evidence-Based Management of Sickle Cell Disease , Expert Panel Report 2014, is based on the best available but limited evidence. When high-quality evidence was lacking, expert consensus was used to provide recommendations. The guideline development methodology is described in detail in the full report. 1
Some groups have used the guidelines to develop implementation strategies, which are presented as information without review or endorsement by the Expert Panel.
Suggestions from Community Care of North Carolina: https://www.communitycarenc.org/chacc-hematology-guidelines/
Summary of SCD guidelines from the National Heart, Lung, and Blood Institute (Quick Guide): http://www.nhlbi.nih.gov/sites/www.nhlbi.nih.gov/files/quickguide.pdf
The views expressed in this article are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute or the U.S. Department of Health and Human Services.
The authors thank George R. Buchanan, MD, and Lanetta Jordan, MD, MPH, MSPH, for assistance with the manuscript. They also thank the following panel members: George R. Buchanan, MD; Araba N. Afenyi-Annan, MD, MPH; Samir K. Ballas, MD; Kathryn L. Hassell, MD; Andra H. James, MD, MPH; Sophie M. Lanzkron, MD, MHS; Richard Lottenberg, MD; William J. Savage, MD, PhD; Paula J. Tanabe, PhD, RN; Russell E. Ware, MD, PhD; M. Hassan Murad, MD, MPH; Jonathan Goldsmith, MD; Eduardo Ortiz, MD, MPH; and Robinson Fulwood, PhD, MSPH.
National Heart Lung, and Blood Institute. Evidence-based management of sickle cell disease. Expert panel report, 2014. http://www.nhlbi.nih.gov/sites/www.nhlbi.nih.gov/files/sickle-cell-disease-report.pdf . Accessed September 29, 2015.
Connes P, Lamarre Y, Waltz X, et al. Haemolysis and abnormal haemorheology in sickle cell anaemia. Br J Haematol. 2014;165(4):564-572.
Ballas SK, Mohandas N. Sickle red cell microrheology and sickle blood rheology. Microcirculation. 2004;11(2):209-225.
Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337(11):762-769.
Brawley OW, Cornelius LJ, Edwards LR, et al. National Institutes of Health Consensus Development Conference statement: hydroxyurea treatment for sickle cell disease. Ann Intern Med. 2008;148(12):932-938.
Haywood C, Beach MC, Lanzkron S, et al. A systematic review of barriers and interventions to improve appropriate use of therapies for sickle cell disease. J Natl Med Assoc. 2009;101(10):1022-1033.
Miller ST, Wright E, Abboud M, et al.; STOP Investigators. Impact of chronic transfusion on incidence of pain and acute chest syndrome during the Stroke Prevention Trial (STOP) in sickle-cell anemia. J Pediatr. 2001;139(6):785-789.
Raphael JL, Oyeku SO, Kowalkowski MA, Mueller BU, Ellison AM. Trends in blood transfusion among hospitalized children with sickle cell disease. Pediatr Blood Cancer. 2013;60(11):1753-1758.
Gaston MH, Verter JI, Woods G, et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N Engl J Med. 1986;314(25):1593-1599.
Falletta JM, Woods GM, Verter JI, et al. Discontinuing penicillin prophylaxis in children with sickle cell anemia. Prophylactic Penicillin Study II. J Pediatr. 1995;127(5):685-690.
Nkouwap I, Diara JP, Noyon I, Étienne-Julan M, Mérault L. Is there any alternative to oral penicillin in antibioprophylaxis for children with sickle cell disease? [French]. Med Mal Infect. 1999;29(2):111-116.
Hussain S, Nichols F, Bowman L, Xu H, Neunert C. Implementation of transcranial Doppler ultrasonography screening and primary stroke prevention in urban and rural sickle cell disease populations [published online ahead of print November 8, 2104]. Pediatr Blood Cancer . http://onlinelibrary.wiley.com/doi/10.1002/pbc.25306/abstract;jsessionid=29B91A94C6C6DF5DD715B5C97694E336.f01t04 . Accessed September 22, 2015.
Goldstein LB, Adams R, Becker K, et al. Primary prevention of ischemic stroke: A statement for healthcare professionals from the Stroke Council of the American Heart Association. Circulation. 2001;103(1):163-182.
Enninful-Eghan H, Moore RH, Ichord R, Smith-Whitley K, Kwiatkowski JL. Transcranial Doppler ultrasonography and prophylactic transfusion program is effective in preventing overt stroke in children with sickle cell disease. J Pediatr. 2010;157(3):479-484.
Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339(1):5-11.
Adams RJ, Brambilla D Optimizing Primary Stroke Prevention in Sickle Cell Anemia (STOP 2) Trial Investigators. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. N Engl J Med. 2005;353(26):2769-2778.
Smith-Whitley K. Reproductive issues in sickle cell disease. Blood. 2014;124(24):3538-3543.
Centers for Disease Control and Prevention. U.S. medical eligibility criteria for contraceptive use, 2010. MMWR Recomm Rep. 2010;59(RR-4):1-6.
Barfield WD, Barradas DT, Manning SE, Kotelchuck M, Shapiro-Mendoza CK. Sickle cell disease and pregnancy outcomes: women of African descent. Am J Prev Med. 2010;38(4 suppl):S542-S549.
Alayed N, Kezouh A, Oddy L, Abenhaim HA. Sickle cell disease and pregnancy outcomes: population-based study on 8.8 million births. J Perinat Med. 2014;42(4):487-492.
Johnson K, Posner SF, Biermann J, et al. Recommendations to improve preconception health and health care—United States. A report of the CDC/ATSDR Preconception Care Work Group and the Select Panel on Preconception Care. MMWR Recomm Rep. 2006;55(RR-6):1-23.
Smith WR, Penberthy LT, Bovbjerg VE, et al. Daily assessment of pain in adults with sickle cell disease. Ann Intern Med. 2008;148(2):94-101.
Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med. 1991;325(1):11-16.
Ballas SK, Gupta K, Adams-Graves P. Sickle cell pain: a critical reappraisal. Blood. 2012;120(18):3647-3656.
Jacobson SJ, Kopecky EA, Joshi P, Babul N. Randomised trial of oral morphine for painful episodes of sickle-cell disease in children. Lancet. 1997;350(9088):1358-1361.
Hagmeyer KO, Mauro LS, Mauro VF. Meperidine-related seizures associated with patient-controlled analgesia pumps. Ann Pharmacother. 1993;27(1):29-32.
Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members [published corrections appear in JAMA . 2014;312(18):1932, and JAMA . 2015;313(7):729]. JAMA. 2014;312(10):1033-1048.
Vichinsky EP, Neumayr LD, Earles AN, et al.; National Acute Chest Syndrome Study Group. Causes and outcomes of the acute chest syndrome in sickle cell disease [published correction appears in N Engl J Med . 2000;343(11):824]. N Engl J Med. 2000;342(25):1855-1865.
Abbas HA, Kahale M, Hosn MA, Inati A. A review of acute chest syndrome in pediatric sickle cell disease. Pediatr Ann. 2013;42(3):115-120.
Ahmad FA, Macias CG, Allen JY. The use of incentive spirometry in pediatric patients with sickle cell disease to reduce the incidence of acute chest syndrome. J Pediatr Hematol Oncol. 2011;33(6):415-420.
Booth C, Inusa B, Obaro SK. Infection in sickle cell disease: a review. Int J Infect Dis. 2010;14(1):e2-e12.
Charache S, Terrin ML, Moore RD, et al.; Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. N Engl J Med. 1995;332(20):1317-1322.
Rodgers GP, Dover GJ, Noguchi CT, Schechter AN, Nienhuis AW. Hematologic responses of patients with sickle cell disease to treatment with hydroxyurea. N Engl J Med. 1990;322(15):1037-1045.
Platt OS, Orkin SH, Dover G, Beardsley GP, Miller B, Nathan DG. Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. J Clin Invest. 1984;74(2):652-656.
Wang WC, Ware RE, Miller ST, et al.; BABY HUG investigators. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet. 2011;377(9778):1663-1672.
Wang WC, Wynn LW, Rogers ZR, Scott JP, Lane PA, Ware RE. A two-year pilot trial of hydroxyurea in very young children with sickle-cell anemia. J Pediatr. 2001;139(6):790-796.
Kinney TR, Helms RW, O'Branski EE, et al.; Pediatric Hydroxyurea Group. Safety of hydroxyurea in children with sickle cell anemia: results of the HUG-KIDS study, a phase I/II trial. Blood. 1999;94(5):1550-1554.
Steinberg MH, McCarthy WF, Castro O, et al.; Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia and MSH Patients' Follow-Up. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: a 17.5 year follow-up. Am J Hematol. 2010;85(6):403-408.
Chou ST. Transfusion therapy for sickle cell disease: a balancing act. Hematology Am Soc Hematol Educ Program. 2013;2013:439-446.
Smith-Whitley K, Thompson AA. Indications and complications of transfusions in sickle cell disease. Pediatr Blood Cancer. 2012;59(2):358-364.
Continue Reading

More in AFP
Copyright © 2015 by the American Academy of Family Physicians.
This content is owned by the AAFP. A person viewing it online may make one printout of the material and may use that printout only for his or her personal, non-commercial reference. This material may not otherwise be downloaded, copied, printed, stored, transmitted or reproduced in any medium, whether now known or later invented, except as authorized in writing by the AAFP. See permissions for copyright questions and/or permission requests.
Copyright © 2024 American Academy of Family Physicians. All Rights Reserved.
What is Sickle Cell Disease?

Sickle cell disease (SCD) is a group of inherited red blood cell disorders. Red blood cells contain hemoglobin, a protein that carries oxygen. Healthy red blood cells are round, and they move through small blood vessels to carry oxygen to all parts of the body. In someone who has SCD, the hemoglobin is abnormal, which causes the red blood cells to become hard and sticky and look like a C-shaped farm tool called a “sickle.” The sickle cells die early, which causes a constant shortage of red blood cells. Also, when they travel through small blood vessels, they get stuck and clog the blood flow. This can cause pain and other serious complications (health problems) such as infection, acute chest syndrome and stroke.
Types of SCD
There are several types of SCD. The specific type of SCD a person has depends on the genes they inherited from their parents. People with SCD inherit genes that contain instructions, or code, for abnormal hemoglobin.
Below are the most common types of SCD:
People who have this form of SCD inherit two genes, one from each parent, that code for hemoglobin “S.” Hemoglobin S is an abnormal form of hemoglobin that causes the red cells to become rigid, and sickle shaped. This is commonly called sickle cell anemia and is usually the most severe form of the disease.
People who have this form of SCD inherit a hemoglobin “S” gene from one parent and a gene for a different type of abnormal hemoglobin called “C” from the other parent. This is usually a milder form of SCD.

Did you know SCD affects people from many parts of the world?
HbS beta thalassemia
People who have this form of SCD inherit a hemoglobin “S” gene from one parent and a gene for beta thalassemia, another type of hemoglobin abnormality, from the other parent. There are two types of beta thalassemia: “zero” (HbS beta 0 ) and “plus” (HbS beta + ). Those with HbS beta 0 -thalassemia usually have a severe form of SCD. People with HbS beta + -thalassemia tend to have a milder form of SCD.
There also are a few rare types of SCD, such as the following:
Hbsd, hbse, and hbso.
People who have these forms of SCD inherit one hemoglobin “S” gene and one gene that codes for another abnormal type of hemoglobin (“D”, “E”, or “O”). The severity of these rarer types of SCD varies.
Sickle Cell Trait (SCT)
People who have sickle cell trait (SCT) inherit a hemoglobin “S” gene from one parent and a normal gene (one that codes for hemoglobin “A”) from the other parent. People with SCT usually do not have any of the signs of the disease. However, in rare cases, a person with SCT may develop health problems; this occurs most often when there are other stresses on the body, such as when a person becomes dehydrated or exercises strenuously. Additionally, people who have SCT can pass the abnormal hemoglobin “S” gene on to their children.
Learn more about sickle cell trait »
Cause of SCD
SCD is a genetic condition that is present at birth. It is inherited when a child receives two genes—one from each parent—that code for abnormal hemoglobin.
SCD is diagnosed with a simple blood test. In children born in the United States, it most often is found at birth during routine newborn screening tests at the hospital. In addition, SCD can be diagnosed while the baby is in the womb. Diagnostic tests before the baby is born, such as chorionic villus sampling and amniocentesis , can check for chromosomal or genetic abnormalities in the baby. Chorionic villus sampling tests a tiny piece of the placenta, called chorionic villus. Amniocentesis tests a small sample of amniotic fluid surrounding the baby.
Because children with SCD are at an increased risk of infection and other health problems, early diagnosis and treatment are important.
Talk to your doctor to find out how to get tested and to explain the results after testing .
View and print
Complications
People with SCD may start to have signs of the disease during the first year of life, usually around 5 months of age. Symptoms and complications of SCD are different for each person and can range from mild to severe. Learn about the complications.
Prevention and Treatment of SCD Complications
General Prevention Strategies
Management of SCD is focused on preventing and treating pain episodes and other complications. Prevention strategies include lifestyle behaviors as well as medical screening and interventions to prevent SCD complications.
Lifestyle Behaviors
There are simple steps that people with SCD can take to help prevent and reduce the occurrence of pain crises, including the following:
- Drink plenty of water.
- Try not to get too hot or too cold.
- Try to avoid places or situations that cause exposure to high altitudes (for example, flying, mountain climbing, or cities with a high altitude).
- Try to avoid places or situations with exposure to low oxygen levels (for example, mountain climbing or exercising extremely hard, such as in military boot camp or when training for an athletic competition).
Simple steps to prevent harmful infections include the following:
- Wash your hands often . Washing hands with soap and clean water many times each day is one of the best ways people with SCD, their family members, and other caregivers can help prevent an infection.
- Prepare food safely . Bacteria can be especially harmful to children with SCD.
Medical Screenings & Interventions to Prevent SCD Complications
Prevention of Infections
- Vaccines can protect against harmful infections. It is important that children with SCD get all regular childhood vaccines . Similarly, it is important for children and adults to get the flu vaccine every year, as well as the pneumococcal vaccine and any others recommended by a doctor.
- Penicillin greatly reduces the risk of infections in people with HbSS and has been shown to be even more effective when it is started earlier. To decrease the risk of infection, it’s important that young children with HbSS take penicillin (or other antibiotic prescribed by a doctor) every day until at least 5 years of age. Penicillin on a daily basis is usually not prescribed for children with other types of SCD unless the severity of the disease is similar to that of HbSS, such as HbS beta 0 -thalassemia.
Prevention of Vision Loss
- Yearly visits to an eye doctor to look for damage to the retina (the part of your eye that senses light and sends images to your brain) are important for people with SCD to avoid vision loss. If possible, it’s best to see an eye doctor who specializes in diseases of the retina.
- If the retina is damaged by excessive blood vessel growth, laser treatment often can prevent further vision loss.
Prevention of Stroke
- Children who are at risk for stroke can be identified using a special type of exam called transcranial Doppler ultrasound (TCD). If the child is found to have an abnormal TCD, a doctor might recommend frequent blood transfusions (a procedure in which new blood is put into a person’s body through a small plastic tube inserted into a person’s blood vessels) to help prevent a stroke.
- People who have frequent blood transfusions are usually watched closely because there can be serious side effects. For example, because blood contains iron, transfusions can lead to a condition called iron overload, in which too much iron builds up in the body. Iron overload can cause life-threatening damage to the liver, heart, and other organs. Therefore, it is important for people with SCD receiving regular blood transfusions to also receive treatment to reduce excess iron in the body. This type of treatment is known as iron chelation therapy.
Prevention of Severe Anemia
- Blood transfusions may be used to treat severe anemia. A sudden worsening of anemia resulting from infection or enlargement of the spleen (an organ in the upper left side of the abdomen) is a common reason for a transfusion.
- As with preventing stroke, frequent blood transfusions can cause iron overload, and iron chelation therapy may be needed to reduce excess iron in the body.
Management of Pain Crises
When pain crises do occur, clinical management may include the following:
- Intravenous fluids (giving fluids directly into a person’s vein)
- Pain-reducing medicine
- Hospitalization for severe pain crises
Specific Treatments to Prevent SCD Complications
SCD is a disease that worsens over time. Treatments are available that can prevent complications and lengthen the lives of those who have this condition. These treatment options and their effects can be different for each person, depending on the symptoms and severity of their disease. It is important to understand the benefits and risks of each treatment option. Currently, the FDA has approved four treatments for SCD [1] .
- Hydroxyurea (pronounced “hi-DROK-see-yoo-REE-uh”) may help people with SCD ages 2 years and older. More information about hydroxyurea can be found here .
- L-glutamine (pronounced “L-gloo-ta-meen,”), or ENDARI® may help people with SCD ages 5 years and older. More information about L-glutamine can be found on page 2 here .
- Voxelotor (pronounced “vox-EL-o-tor”), or OXBRYTA® may help people with SCD ages 4 years and older. More information about Voxelotor can be found on the FDA website here .
- Crizanlizumab (pronounced “criz-an-liz-u-mab”), or ADAKVEO® may help people with SCD ages 16 years and older. More information about Crizanlizumab can be found on page 1 here .
Several other treatments and therapies for SCD have recently been developed that are still undergoing clinical trials and thus have not yet been approved by the FDA.
The only therapy approved by the FDA that may be able to cure SCD is a bone marrow or stem cell transplant.
Bone marrow is a soft, fatty tissue inside the center of the bones, where blood cells are made. A bone marrow or stem cell transplant is a procedure that takes healthy cells that form blood from one person—the donor—and puts them into someone whose bone marrow is not working properly.
Bone marrow or stem cell transplants are very risky and can have serious side effects, including death. For the transplant to work, the bone marrow must be a close match. Usually, the best donor is a brother or sister. Bone marrow or stem cell transplants are most common in cases of severe SCD for children who have minimal organ damage from the disease.
Find clinical treatment guidelines for providers as well as patient and family resources derived from clinical treatment guidelines:
For healthcare providers.
- Evidence-Based Management of Sickle Cell Disease: Expert Panel Report, 2014, National Heart, Lung, and Blood Institute
- Clinical Practice Guidelines on Sickle Cell Disease, American Society of Hematology
For People with SCD and their Families
- Steps to Better Health for People with Sickle Cell Disease Toolkit
[1] CDC will periodically review and update this treatment list when new treatments are approved by the FDA.

Join the Public Health Webinar Series on Blood Disorders
- Upcoming webinar
- View past webinars
Exit Notification / Disclaimer Policy
- The Centers for Disease Control and Prevention (CDC) cannot attest to the accuracy of a non-federal website.
- Linking to a non-federal website does not constitute an endorsement by CDC or any of its employees of the sponsors or the information and products presented on the website.
- You will be subject to the destination website's privacy policy when you follow the link.
- CDC is not responsible for Section 508 compliance (accessibility) on other federal or private website.
IMAGES